【4.2.1】序列比对到参考基因组-- hisat2
一、 基本介绍
HISAT2 是一款利用改进的BWT算法进行序列比对的软件。由约翰霍普金斯大学计算生物学中心(CCB at JHU)开发,是TopHat的升级版本,速度提高了50倍。利用 HISAT2 + StringTie 流程,可以快速地分析转录组测序数据,获得每个基因和转录本的表达量。
- TopHat采用外显子优先的方法,第一步使用BWT方法将完整的匹配外显子的reads优先回贴到参考基因组,第二步将没有比对到基因组的reads切割成小片段,分别比对到基因组。
- HISAT2使用了叠加的比对算法,全局比对整个基因组建立index,辅助成千上万个小的局部index。
- STAR采用了种子和扩展的方法。
首先需要构建参考基因组索引用于下一步的比对。HISAT2提供了两个脚本用于从基因组注释GTF文件中提取剪接位点和外显子位置,基于这些特征,可以使 RNA-Seq reads 比对更加准确。建议直接在HISAT2官网下载构建好的index。(为什么需要索引:为了提高比对速度,需要根据参考基因组序列,经过BWT算法转换成index,而我们比对的序列其实是index的一个子集。因此不是直接把reads回贴到基因组上,而是把reads与index进行比较。)
然后再进行reads mapping。reads比对到基因组上的一个位置称为一个alignment。软件会对所有的alignments进行打分和判断,能有符合过滤条件的valid alignment才会输出。打分机制包括:错配碱基罚分(–mp),reads上的gap罚分(–rdg),reference上的gap罚分(–rdg),valid alignment分数阈值(–score—min,默认L,0,-0.2),多个valid alignments输出数目(-k,默认5)。
二、 背景知识
(1) BWT索引
处理NGS测序数据最常见的任务(task)是读段映射(read mapping):在参考基因组序列(reference genomic sequence)中定位读段(locating reads)并计算相应的比对(alignments)。在这里,输入的reads通常来自正在研究的有机体(例如患病个体),参考基因组是已知的具有代表性的物种基因组序列(例如参考人类基因组序列),或可能是系统发育相关的物种(phylogenetically related species)。一个主要的困难来自数据的大小:数以百万计的reads必须与一个可以包含数十亿个字母的序列进行比对。类似BLAST的工具根本无法在合理的时间内处理这样的数据。
开发出了各种各样的read mapping软件。最常见的实用映射软件包括BWA-MEM、Bowtie2、GEM,它们比类似BLAST的工具快几个数量级。这些算法的一个主要效率(efficiency)来源是被称为BWT-或FM-index的索引结构(indexing structure)。
BWT-index的根源在于词组合(word combinatorics):它基于Burrows-Wheeler变换(Burrows-Wheeler transform),从组合的角度来看,它与Gessel-Reutenauer双射(bijection,一一对应关系)有着密切的联系。虽然BWT的主要应用是文本压缩(text compression),但它被应用于构造一个简洁的文本索引(compact text index),结果令人惊讶地强大。
与以BLAST为代表的基于哈希的索引基础(hash-based indexes underpinning)支持种子和扩展策略(seed-and-extend strategies)不同,BWT索引是一个全文索引(full-text index),因为它支持搜索任意大小的字符串(arbitrary-size strings)。BWT-index属于一个更普遍的简洁索引(succinct indexes)系列,其以位(bits)为单位的大小接近于对象(这里是序列)的无损表示(lossless representation of the object)所需的信息理论最小值(information-theoretic minimum)。 ——Evolution of biosequence search algorithms: a brief survey
三、 hisat2使用方法
(1) 用法和参数
hisat2 [options]* -x <ht2-idx> {-1 <m1> -2 <m2> | -U <r>} [-S <sam>]
#提取外显子和剪接位点
extract_splice_sites.py dmel.annotation.gtf > dmel.ss
extract_exons.py dmel.annotation.gtf > dmel.exon
extract_snps.py snp142Common.txt > genome.snp
#构建参考基因组索引
hisat2-build --ss dmel.ss --exon dmel.exon (--snp genome.snp) dmel.genome.fa dmel
#得到8个索引文件,以dmel为共同的前缀,dmel.1~8.ht2
# Reads mapping
hisat2 -p 4 --dta -x dmel -1 sample_1.fq.gz -2 sample_2.fq.gz -S sample.sam
{-1 <m1> -2 <m2> | -U <r>}:对于单端数据,采用-U指定输入文件(-U <r>);对于双端数据,采用-1和-2分别指定R1端和R2端的输入文件(-1 <m1> -2 <m2>)。
-x:指定基因组索引的文件前缀(前缀.1~8.ht2)
-S:指定输出的SAM文件<sam>。默认输出到标准输出,比对结束后统计结果输出到标准错误输出。
-p:线程数。
--dta:输出专门为转录本组装的比对结果。Reports alignments tailored for transcript assemblers.(如果比对结果后续需要使用StringTie进行转录本组装,则一定要加入--dta选项。)
--rna-strandness:默认unstranded,如果是链特异性文库要加上--rna-strandness RF。
-f:表示输入文件格式为fasta(默认),-q表示输入文件格式为fastq。可以是gzip压缩的文件。
-phred33:输入的FASTQ文件碱基质量值编码标准为phred33(默认)。
-k <int>:report up to <int> alignments per read; MAPQ not meaningful.
(2) 举个例子
#提取外显子和剪接位点
extract_splice_sites.py annotation/genome/Drosophila_melanogaster.BDGP6.32.105.gtf > annotation/genome/dmel.ss
extract_exons.py annotation/genome/Drosophila_melanogaster.BDGP6.32.105.gtf > annotation/genome/dmel.exon
#构建参考基因组索引
hisat2-build --ss annotation/genome/dmel.ss --exon annotation/genome/dmel.exon annotation/genome/Drosophila_melanogaster.BDGP6.32.dna.toplevel.fa ./annotation/index/dmel 2>>log/hisat2_index_log.txt & #在annotation/index/文件夹下生成了8个索引文件
# Reads mapping
hisat2 -p 4 --dta --rna-strandness RF -x ./annotation/index/dmel -1 02_cleandata/WTF4.Input_1_val_1.fq.gz -2 02_cleandata/WTF4.Input_2_val_2.fq.gz -S 04_bam/WTF4.Input.sam 2>>log/hisat2_log.txt #双端,链特异性文库
# SAM转为BAM
samtools sort -@ 4 -o 04_bam/WTF4.Input.sorted.bam 04_bam/WTF4.Input.sam
(3) 批量处理
#以双端为例:
for i in `ls 02_cleandata/*_1_val_1.fq.gz `
do
i=`basename $i`
i=${i%*_1_val_1.fq.gz}
echo $i >>log/hisat2_log.txt
hisat2 -p 4 --dta --rna-strandness RF -x ./annotation/index/dmel -1 02_cleandata/$i\_1_val_1.fq.gz -2 02_cleandata/$i\_2_val_2.fq.gz 2>>log/hisat2_log.txt | samtools sort -@ 4 -o 04_bam/$i.bam 2>>log/hisat2_log.txt
done
(4) 质控和建立索引
#批量对bam文件进行QC
ls 04_bam/*.bam | while read id ; do (samtools flagstat $id > 04_bam/$(basename $id ".bam").stat) ; done &
#每一个bam文件都生成了一个stat文件
274 + 0 in total (QC-passed reads + QC-failed reads)
50 + 0 primary
224 + 0 secondary
0 + 0 supplementary
0 + 0 duplicates
0 + 0 primary duplicates
270 + 0 mapped (98.54% : N/A)
46 + 0 primary mapped (92.00% : N/A)
50 + 0 paired in sequencing
25 + 0 read1
25 + 0 read2
46 + 0 properly paired (92.00% : N/A)
46 + 0 with itself and mate mapped
0 + 0 singletons (0.00% : N/A)
0 + 0 with mate mapped to a different chr
0 + 0 with mate mapped to a different chr (mapQ>=5)
#批量对bam文件建立索引
for i in `ls 04_bam/*.bam` ; do (samtools index $i) ; done &
#每一个bam文件都生成了一个bam.bai文件
(5) 结果解读
通过2»log将比对率等信息的标准错误输出重定向到log文件。
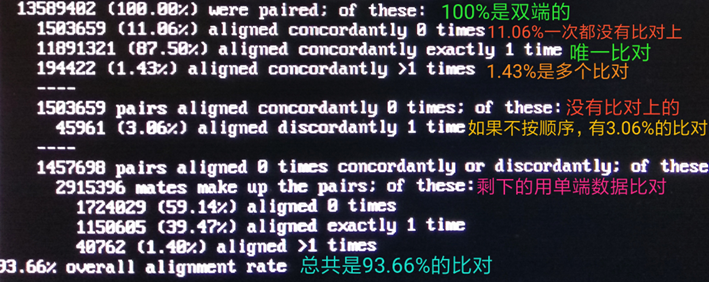
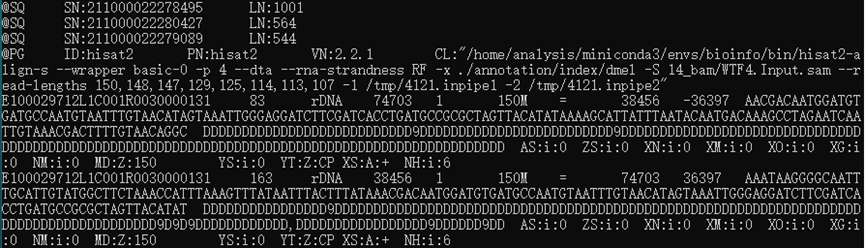
查看SAM结果:less 04_bam/WTF4.Input.sam
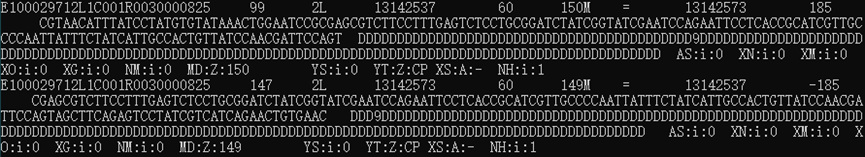
查看BAM结果:samtools view 04_bam/WTF4.Input.sorted.bam | less
四、我的案例
hisat2 --no-spliced-alignment --no-unal -x /opt/ref/ref_idx -1 ${qc1} -2 ${qc2} --rg-id ${ID} --rg SM:${ID} --rg LB:Illumina -q | samtools view -Sb -F256 - | samtools sort > ${output}/${ID}/${ID}.bam
参考资料
