【3.3.1】IGV –基因组可视化工具
IGV(Integrative Genomics Viewer)是一个基因组可视化的工具
一、简介
官网:http://www.broadinstitute.org/igv/
使用说明:http://www.broadinstitute.org/igv/book/export/html/6
下载地址:http://www.broadinstitute.org/software/igv/download
学习资料:http://pan.baidu.com/s/1o8SDf42
我选择的版本 Binary Distribution
解压缩后,在cmd终端中输入
e:
cd e:\backup\igv_2.3.67
java -Xmx750m -jar igv.jar
(可以调整750的数值来改变IGV可调用的内存)
运行方式二:
sh igv.sh
在igv.sh中调节内存的大小
二、使用说明
更多内容参见:
https://mp.weixin.qq.com/s/b0FC0wGdDC3nenj2a7bicQ
2.1 数据的导入
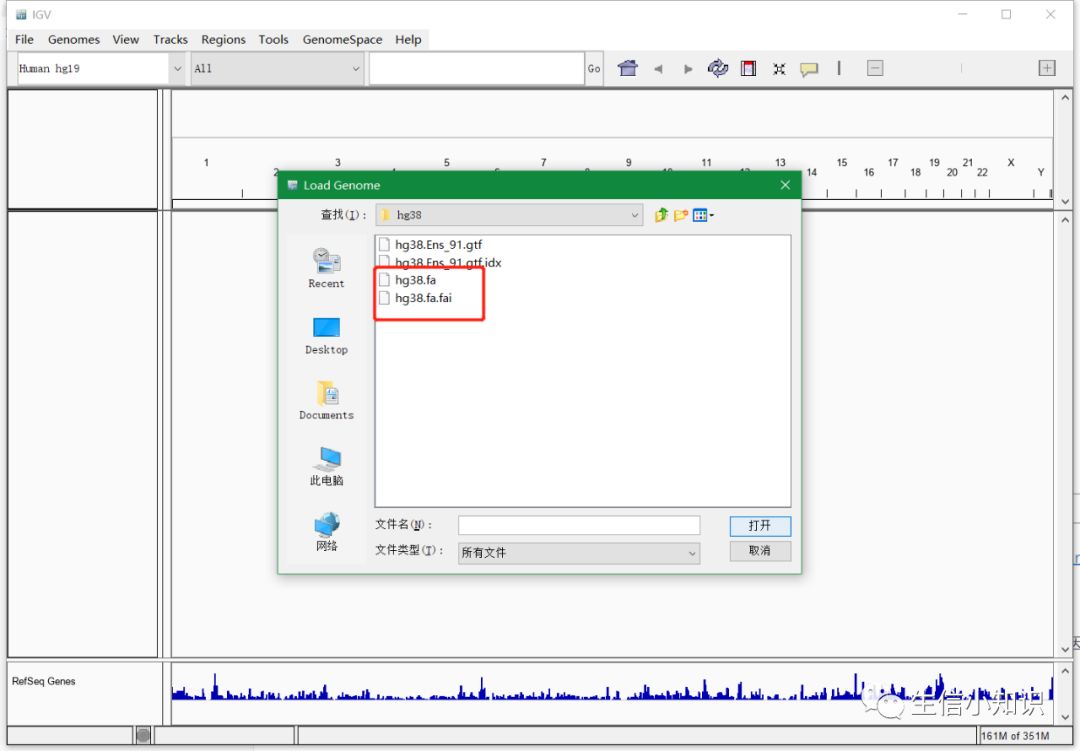
导入基因组文件:(这里是直接导入自己下载的基因组序列fa文件)
在最上面的选项中依次点击:
Genomes→load genomes from file→选择基因组文件(fa文件,注意:一定要有对应的index文件,也就是图中.fai结尾的文件)
如下图:
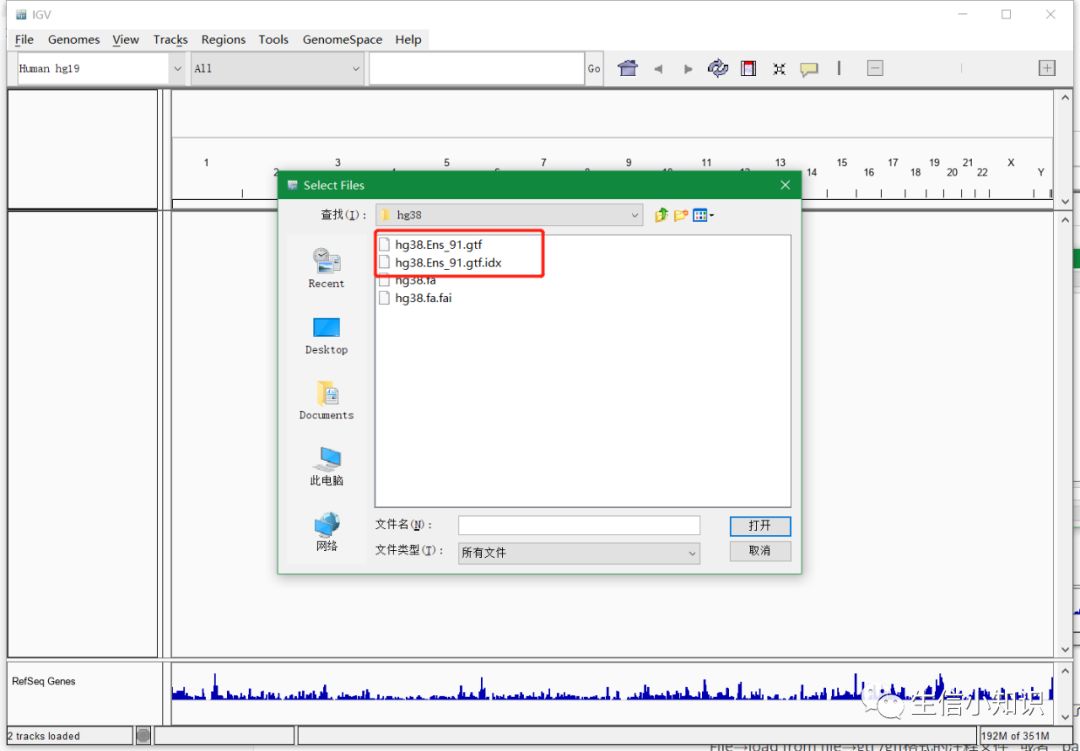
导入基因注释文件和需要查看的目的bam文件:
导入这两种文件的步骤是相同的,故这里直接写在一起了:
File→load from file→gtf /gff格式的注释文件 或者 bam文件
需要注意:这里的注释文件和bam文件都需要对其做index!!(bam的index文件以.bai结尾,注释文件以.idx结尾)
如下图:
2.2 设置中心线帮助浏览
先说一个很方便实用的小工具:
我们放大缩小视野及移动视野的功能主要通过工具栏中的滑动按钮和标尺区域实现,具体的用法就不介绍了,在这里我主要介绍如何在放大过程中始终让目标区域处在视野正中央的位置,而不会随着放大走出视野。
这个技巧对于浏览大型基因组时很有用。这项功能主要通过中心线(center line)实现,如图中红框和红箭头所示:
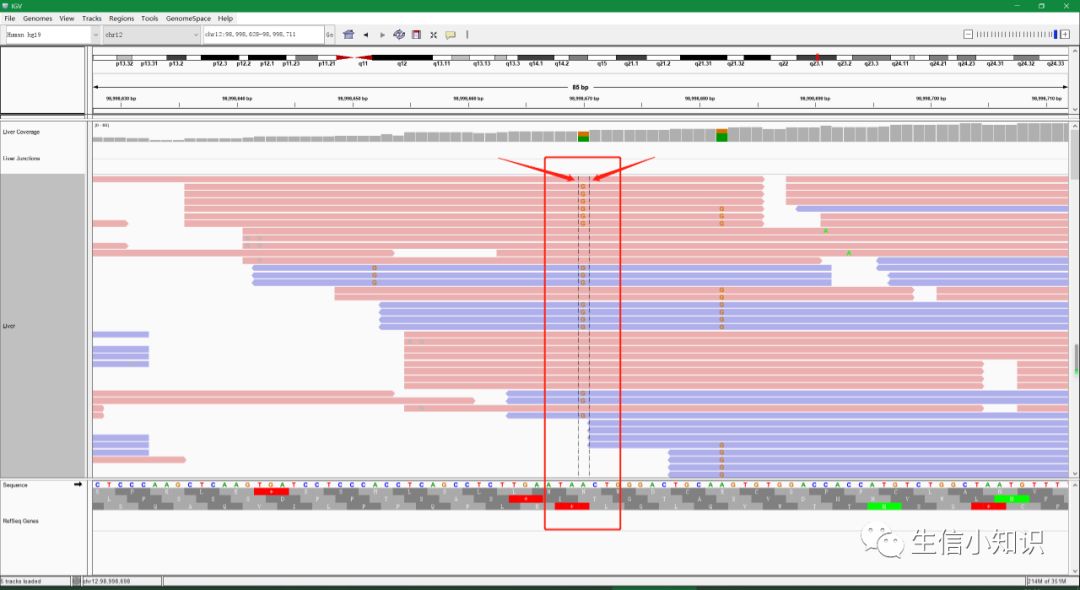
我们可以把想看的区域移动到中心线,然后点击放大,这样想看的区域就不会向左或向右移动脱离视野。
设置中心线方法:主菜单View→Preferences→alignments→show center line

2.3 保存目标区域
:选择一个区域并将其存为感兴趣的区域。
具体用法:
点击这个按钮后,分别在不同的坐标点击2次,第一次代表起点坐标,第二次代表终止坐标。该区域就被存储为一个感 兴趣的区域。点击主菜单中的 Regions→Regions→Navigator,我们可以进入一个关 于感兴趣区域的列表,在列表中我们可以方便的对区域进行编辑。通过点击 view,视野会 转移到之前所存储的感兴趣区域。
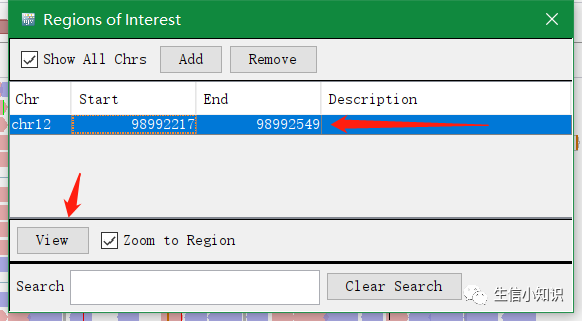
如图所示,这里我保存了一个感兴趣的区域,在经过一段时间的查看后,如果我想再次查看这个位置,那么通过在Navigator里,我可以轻松的返回到这里,非常的方便。
2.4 注释信息的显示方法
:点击这个图标后有3个选项,分别为:
- show details on hover:总是悬浮显示
- show details on click:单击鼠标时显示
- never show details:总不显示
很简单,大家试试就知道了,就不多说了,应该很多人都知道。写在这里只是因为之前我不知道而已。
2.5 辅助线
:点击这个图标后会出现一条辅助线。
同样的,这个功能应该很多人都知道。写在这里只是因为之前我不知道而已。
很多时候这个功能能帮助我们清晰的看懂图。例如:
图中箭头指的黑细线就是辅助线了,可以看到,当我们查看可变剪切事件的时候,就可以用辅助线来帮助我们识别,相信我,比用肉眼来判断好多了,因为我一开始就是肉眼在看,甚至还拿出了直尺去比划,只因为我之前不知道可以直接用这个工具!
2.6 查看序列信息
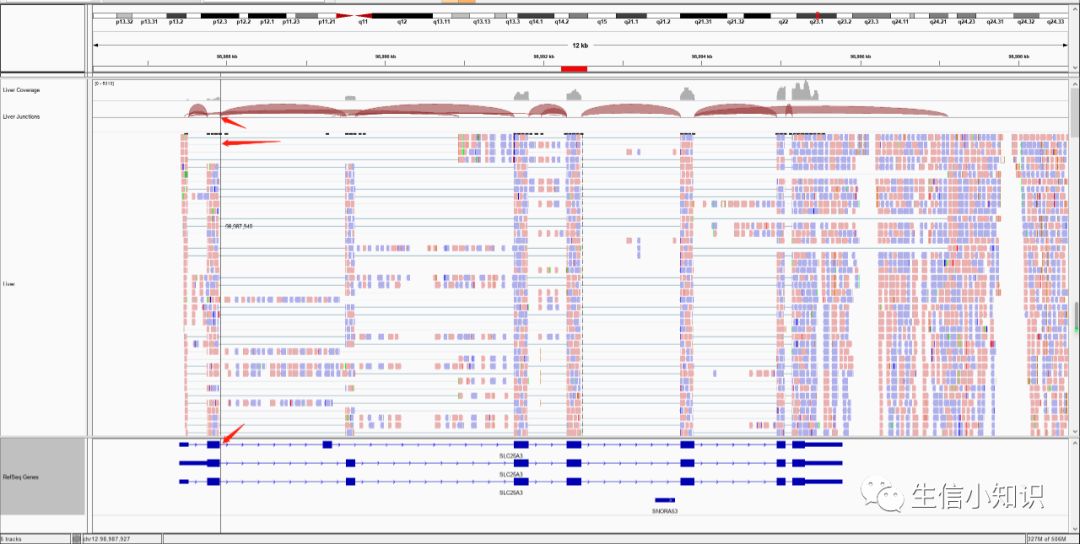
在导入 bam 文件后,我们能在比对结果展示区查看比对相关的信息。如下图所示,我们导入了 Liver 文件后,出现了 3 条 track,Coverage Tack,Junction Track和 Reads Track。
Coverage Tack:主要用于直接展示reads的丰度,高度代表比对到此处的测序片段(read)数目,当比对到参考基因组上的测序片段中有超过 20%与参考基因组不同时,就用不同的颜色标注:红色-T,蓝色-C,绿色-A,橙色-G。否则就用灰色标注。
Junction Track:软件将Read中所检测到的Splice Junction都表示了出来。具体细节后面专门写,详见:sashimi plot。
Reads Track:主要用于直接展示reads的比对情况,具体细节后面专门写,详见:几个重要的右键选项。
2.7 序列信息几个重要方面
在主体图中,实心灰代表比对质量比较高的测序片段,空心灰代表比对到此处的测序片段也可以比对到其他位点。高分辨率下,可以精确到每个位点的碱基类型:当比对序列上与参考基因组相同的超过80%时,用灰色表示;否则用红色-T,蓝色-C,绿色-A,橙色-G,如上图中一些蓝色、绿色、红色及橙色的竖线。
如下图,紫色大写的I字母代表插入(insert),黑色横线代表缺失,鼠标停留在相应位置查看细节。
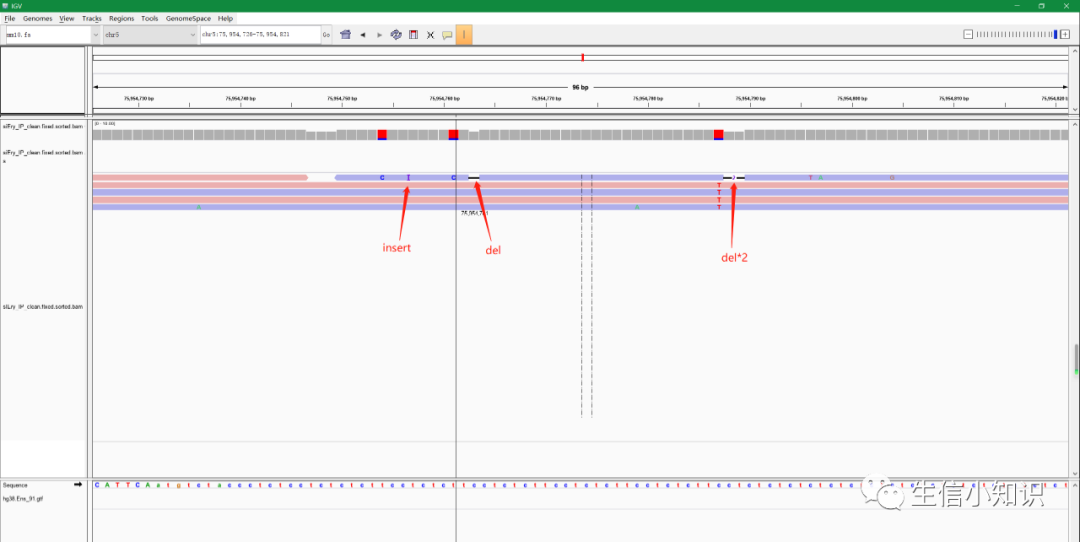
而最上面的那些类似条形图的bar则代表了变异的频率:把鼠标放上去即可看到详细情况

所有的fragment中,如果颜色越浅,则代表着测序质量越差。下图中透明的则代表着测序质量极差,基本不可信。

同理:当放大到一定程度后,每个变异的碱基颜色也有深有浅,浅色的也是测序质量较差的,可信度不高。

2.8 如何查看insert size异常
Insert size是我们寻找可变剪切和融合基因的有利工具,在IGV中我们能通过设定insert size的阈值,来快速识别insert size的异常。首先我们能在右键菜单中的Set insert size options中设置我们需要的阈值,然后在Color alignments by中选择insert size。
完成这些后,视野中超过阈值的的reads会标上不同颜色,蓝色代表insert size比阈值要小,红色代表insert size比阈值要大,其他颜色代表着read比对到另一个染色体上,并且不同颜色代表不同的染色体。
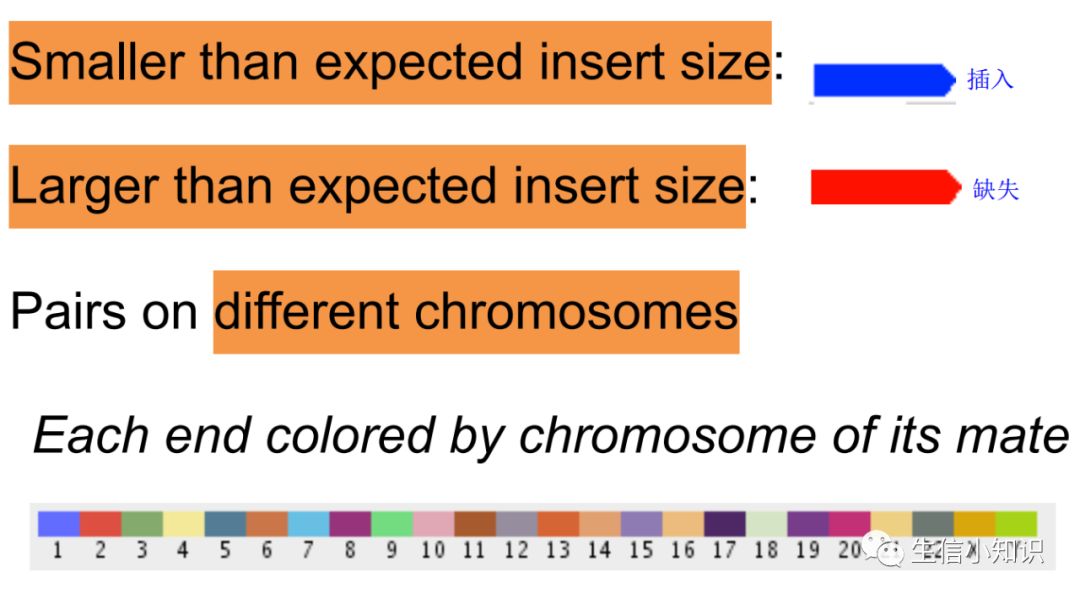
这里其实也很容易理解,如果inferred insert size>expected insert size,也就是说参考基因组中的序列长度比我们测序得到的序列长度要更长,那不就是说明有缺失存在嘛!同理:如果inferred insert size<expected insert size,也就是说参考基因组中的序列长度比我们测序得到的序列长度要更短,那不就是说明有插入存在嘛
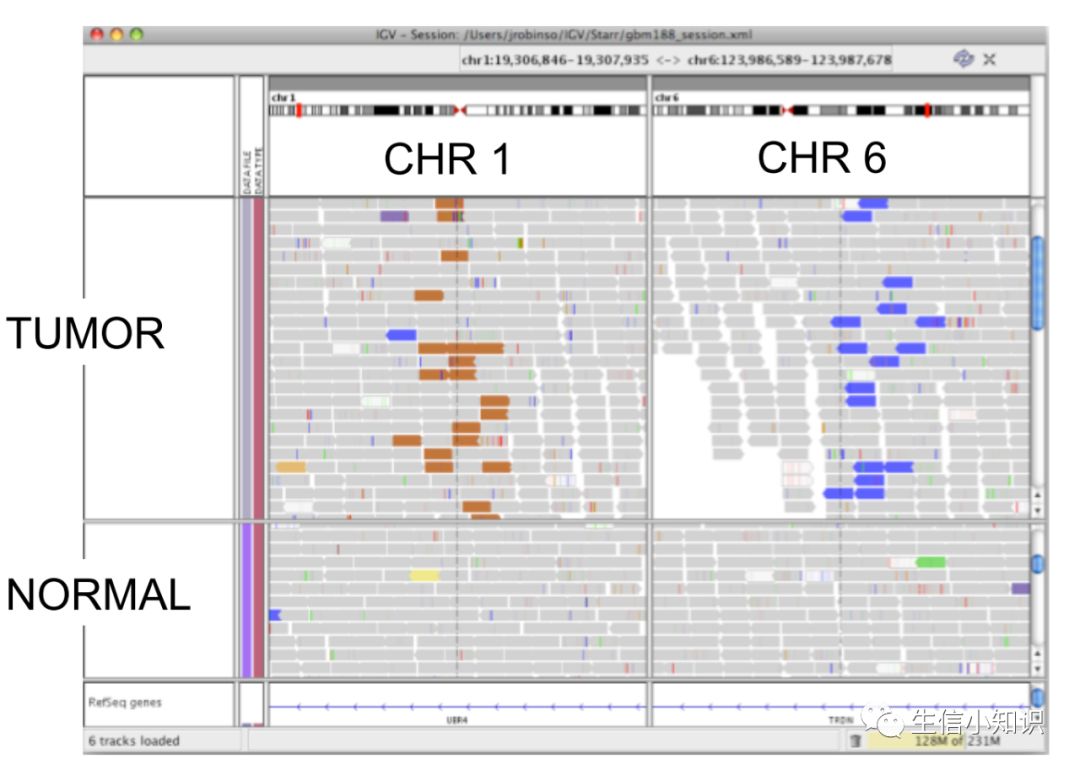
用这个例子来说明下上面讲解的,在肿瘤组织中,1号染色体上有很多棕色的fragment,查表我们知道这个是来自6号染色体的。
同时我们还在6号染色体上也找到了许多蓝色的fragment,说明在6号染色体上还有很多插入存在。
三、讨论
1.Igv使用过程中截图,同时保留深度等信息
PrintScreen + Ctrl C
2.reads红色和蓝色分别代表什么?
IGV uses color coding to flag anomalous insert sizes.
Blue is for inserts that are smaller than expected. That is, the inferred insert size on the reference genome is smaller than expected given the actual insert size.
Red is for inserts that are larger than expected. That is, the inferred insert size on the reference genome is larger than expected given the actual insert size.
蓝色代表插入序列比期望的小
红色代表插入序列比期望的大
3.注意的问题
1.如果上传的是bam或sam文件,需要在同一个文件夹中加入index文件(即.bai)文件 2. 比对结果明明没有插入,但是IGV识别出有插入,应该是bam文件中该序列对应的位置信息不清楚
四、报错
4.1 报错
Error message: 198.bam has invalid uncompressedLength: -1430802916
或者:
invalid gzip header
报错原因:
因为bam文件中,同一个位置的reads数目太多,samtools处理的问题。建议用sambamba来获得bam文件。
参考资料
