【5.2.1.6】COG注释
一 COG简介
COG,即Clusters of Orthologous Groups of proteins。构成每个COG的蛋白都是被假定为来自于一个祖先蛋白,并且因此或者是orthologs或者是paralogs。Orthologs是指来自于不同物种的由垂直家系(物种形成)进化而来的蛋白,并且典型的保留与原始蛋白有相同的功能。Paralogs是那些在一定物种中的来源于基因复制的蛋白,可能会进化出新的与原来有关的功能。请参考文献获得更多的信息。
其网址主页为:http://www.ncbi.nlm.nih.gov/COG/。
网页版使用工具网址:http://www.ncbi.nlm.nih.gov/COG/old/xognitor.html。
使用说明文档网址:http://www.nlm.nih.gov/COG/old/COGhelp.html。
其FTP站点为: ftp://ftp.ncbi.nih.gov/pub/COG/。
通过观看其主页和说明文档,可以理解为COG是NCBI的数据库。COG的中文释义即“同源蛋白簇”。COG分为两类,一类是原核生物的,另一类是真核生物。原核生物的一般称为COG数据库;真核生物的一般称为KOG数据库。
COG是如何建立的?
COG是通过把所有完整测序的基因组的编码蛋白一个一个的互相比较确定的。在考虑来自一个给定基因组的蛋白时,这种比较将给出每个其他基因组的一个最相似的蛋白(因此需要用完整的基因组来定义COG。注1)这些基因的每一个都轮番的被考虑。如果在这些蛋白(或子集)之间一个相互的最佳匹配关系被发现,那么那些相互的最佳匹配将形成一个COG(注2)。这样,一个COG中的成员将与这个COG中的其他成员比起被比较的基因组中的其他蛋白更相像,尽管如果绝对相似性比较的。最佳匹配原则的使用,没有了人为选择的统计切除的限制,这就兼顾了进化慢和进化快的蛋白。然而,还有一个加的限制就是一个COG必须包含来自于3个种系发生上远的基因组的一个蛋白。
COG注释作用:1. 通过已知蛋白对未知序列进行功能注释; 2. 通过查看指定的COG编号对应的protein数目,存在及缺失,从而能推导特定的代谢途径是否存在; 3. 每个COG编号是一类蛋白,将query序列和比对上的COG编号的proteins进行多序列比对,能确定保守位点,分析其进化关系。
二 将序列进行COG分类的步骤
-
COG的ftp里边提供了一个名为myva的文件,该文件里面为COG数据库的蛋白质序列,有192987条。将该序列文件使用使用ncbi-blast-2.2.26+中的blastdb程序制作出一个前缀为cog的蛋白质数据库。
-
将需要进行COG注释并分类的DNA序列或protein序列分别使用blastx或blastp比对到上一步骤建好的cog数据库中。得出xml的比对结果。
-
根据上一步骤的比对结果,得出与query序列相似的cog蛋白id。在COG的ftp里面有一个名为whog的文件,该文件中记录着COG数据中绝大部分的蛋白质id以及其所对应的以COG开头的protein编号,同时也记录这COG编号对应的功能分类编号。因此,可以得出query序列注释的COG编号以及其功能分类编号。
-
ftp中还有一个名为fun.txt的文件,该文件记录这COG的功能分类编号,及其对编号的功能描述。
-
因此,我编写了一个脚本程序用于COG分类注释。该脚本程序名字为cog.pl。输入为所需要进行COG分类的fasta序列文件,得出序列的比对结果和分类统计。
ftp://ftp.ncbi.nih.gov/pub/COG/COG 文件:fun.txt 2 KB 2003/3/2 0:00:00 文件:myva 60217 KB 2003/3/2 0:00:00 文件:myva=gb 3026 KB 2002/9/26 0:00:00 文件:org.txt 4 KB 2003/3/2 0:00:00 文件:pa 769894 KB 2003/3/2 0:00:00 文件:readme 2 KB 2003/4/17 0:00:00 文件:whog 2083 KB 2003/3/5 0:00:00 从03年开始就没有更新了
三 将序列进行KOG分类
方法和COG的分类一致。值得注意的是KOG数据中protein的编号是以LSE或TWOG开头的。同样,我编写了一个kog.pl。
四 说明补充
值得说明的是,KOG数据库中蛋白质序列数目为112920条,但是其中有protein编号的只有27887条,占25%。而COG数据库中蛋白质序列条数为192987,其中有COG编号的有129326条,占67%。所以比对结果中,很多序列比对上了KOG数据库,但是没有protein编号;而在比对到COG数据库时会好很多。 所以,可以先将序列比对到COG数据库,得出分类数据;然后将没有分类编号的序列挑选出来,再比对到KOG数据库,得出分类数据;然后将两个分类数据进行整合,然后画出COG分类图。
五 COG的注释
1,使用Webmga
http://weizhongli-lab.org/metagenomic-analysis
会反馈给你4个结果,output.1和output.2分别是两个非常相近的比对结果,而output.2.family是按照COG的编号排出来的结果,output.2.class是在COG大的分类上的一个统计结果
2,使用陈博士的脚本
mkdir /sam/cog
cd /sam/cog
wget ftp://ftp.ncbi.nih.gov/pub/COG/COG/myva
wget ftp://ftp.ncbi.nih.gov/pub/COG/COG/fun.txt
wget ftp://ftp.ncbi.nih.gov/pub/COG/COG/whog
makeblastdb –in myva –dbtype prot –title cog –parse_seqids –out cog –logfile cog.log
wget http://www.chenlianfu.com/train/bin/cog_from_xml.pl
wget http://www.chenlianfu.com/train/bin/cog_R.pl
使用blast
/sam/blast/bin/blastp -query assembly.orfs.hmm.faa -db /sam/cog /cog -evalue 1e-5 -num_threads 10 -max_target_seqs 5 -outfmt 5 -out assembly.orfs.hmm.blast.xml
-query 输入序列 -db 选择的数据库 -evalue 这个就不说了 -num_threads 服务器使用的cpu个数 -max_target_seqs设置最多的目标序列匹配数 -outfmt输出结果的参数 -out输出的文件名 注意文件或程序的路径,如果没有cd到相应的位置的话,就需要在命令中注明路径
使用cog_from_xml.pl进行COG分类分析
cog_from_xml.pl cog.xml #这一步脚本有问题,做不下去
cog_R.pl cog_cog_class_annot.xls cog_out G.lucidum
display cog_out.png
cog_db_clean.pl -myva myva whog >cog_clean.fa
formatdb -p T -o T -i cog_clean.fa;
3,使用樊博士的脚本
cog_db_clean.pl 并不是所有的COG序列都包含所有的功能注释,所以你需要运行命令,挑选出在COG数据库中有注释的那些序列,脚本下载链接:https://gist.github.com/Buttonwood/96f9a9ef8159ca111a69
cog_db_clean.pl -myva myva whog >cog_clean.fa
# myva 为61.7M的fasta文件,序列名为>AF0017.1这样的格式;cog_clean.fa文件为51M的fasta文件,序列名为>AF0017.1 COG1250 3-hydroxyacyl-CoA dehydrogenase,结合了whog的信息。
formatdb -p T -o T -i cog_clean.fa;
blast_parser.pl 解析blast输出结果,下载链接:https://github.com/JinfengChen/Scripts/tree/master/bin
blastall -p blastp -b 500 -v 500 -F F -d cog_clean.fa -e 1e-4 -i yourdata.fa -o blast.out;
blast_parser.pl -tophit 1 -topmatch 1 blast.out >blast.best;
这个时候你就可以编写perl与R程序输出类似下面的结果:
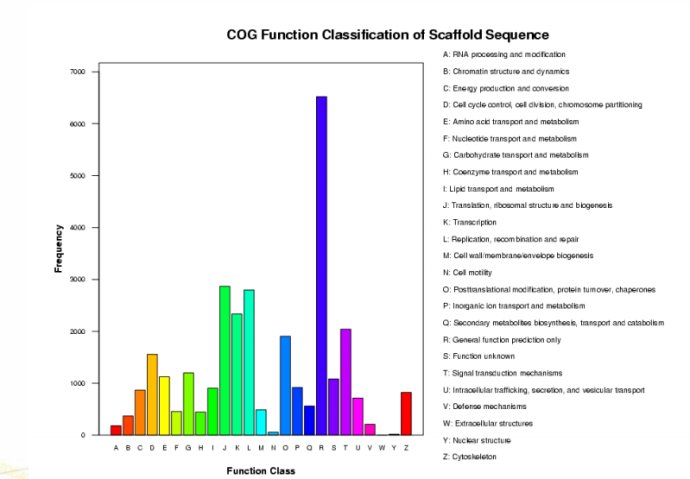
Ps:感谢樊师兄提供的脚本与耐心的解答。
参考资料:
- http://www.chenlianfu.com/?p=366#comments(超赞)
- http://www.biosino.org/pages/ncbi-7.htm
- http://blog.sina.com.cn/s/blog_83f77c940102ux6u.html(超赞)
可以参考的文献:
- Tatusov et al. (1997). A genomic perspective on protein families. Science 278: 631-637. Koonin et al. (1998). Beyond complete genomes: from sequence to structure and function. Curr. Opin. Struct. Biol. 8: 355-363.
- Galperin et al. (1999). Comparing microbial genomes: How the gene set determines the lifestyle. In Organization of the Prokaryotic Genome, R.L. Charlebois, Ed. (American Society of Microbiology, Washington, DC) pp. 91-108.
- Tatusov et al. (2000). A genomic perspective on protein families. Nucleic Acids Res. 28: 33-6.
