【4.2】宏基因组分析--MetaWRAP分析示例
启动:
activate2
conda activate metawrap-env
metaWRAP程序整理了所有的功能模块,可以独立运行。运行metaWRAP -h显示模块名称
Usage: metawrap [module] --help
Options:
read_qc 质控Raw read QC module
assembly 组装Assembly module
binning 分箱Binning module
bin_refinement 分箱提纯Refinement of bins from binning module
reassemble_bins 重装分箱Reassemble bins using metagenomic reads
quant_bins 定量Quantify the abundance of each bin across samples
blobology 可视化Blobology module
kraken 物种注释KRAKEN module
想查看每个模块的具体参数,如组装metawrap assembly -h
Usage: metawrap assembly [options] -1 reads_1.fastq -2 reads_2.fastq -o output_dir
Options:
-1 STR 正向序列forward fastq reads
-2 STR 反向序列reverse fastq reads
-o STR 输出目录output directory
-m INT 内存大小memory in GB (default=10)
-t INT 线程number of threads (defualt=1)
--use-megahit assemble with megahit (default)
--use-metaspades assemble with metaspades instead of megahit
二、具体案例
2.9 下载肠道宏基因组数据
mkdir -p /home/sam/project/metawrap ; cd /home/sam/project/metawrap
##下载3个样品,共6G,解压后15G;包括7G的序列(这里,我只下载了ERR011347这个测序结果)
mkdir raw_data; cd raw_data
wget -c ftp.sra.ebi.ac.uk/vol1/fastq/ERR011/ERR011347/ERR011347_1.fastq.gz;
wget -c ftp.sra.ebi.ac.uk/vol1/fastq/ERR011/ERR011347/ERR011347_2.fastq.gz;
wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR011/ERR011348/ERR011348_1.fastq.gz;
wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR011/ERR011348/ERR011348_2.fastq.gz;
wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR011/ERR011349/ERR011349_1.fastq.gz;
wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR011/ERR011349/ERR011349_2.fastq.gz;
# 解压
gunzip *.gz
# 移动至子目录
mkdir RAW_READS
mv *fastq RAW_READS
2.1 read_qc质控和去宿主
默认使用人类基因组的hg38的bmt索引去宿主,需要在软件安装和数据库配置中提前布置好。对于环境样本,也可以不去宿主,使用 –skip-bmtagger 跳过。
我们对每个样本进行处理
mkdir READ_QC
# 24线程程质控5GB数据,大约29分钟(m)单个样本,但软件好像不支持多线程
time metawrap read_qc -1 RAW_READS/ERR011347_1.fastq -2 RAW_READS/ERR011347_2.fastq -t 24 -o READ_QC/ERR011347
metawrap read_qc -1 RAW_READS/ERR011348_1.fastq -2 RAW_READS/ERR011348_2.fastq -t 24 -o READ_QC/ERR011348
metawrap read_qc -1 RAW_READS/ERR011349_1.fastq -2 RAW_READS/ERR011349_2.fastq -t 24 -o READ_QC/ERR011349
我们可以查看每个样品目录中的报告,来比较质控前后的变化,如READ_QC/ERR011347/pre-QC_report/ERR011347_2_fastqc.html包含质控前的质量评估结果。
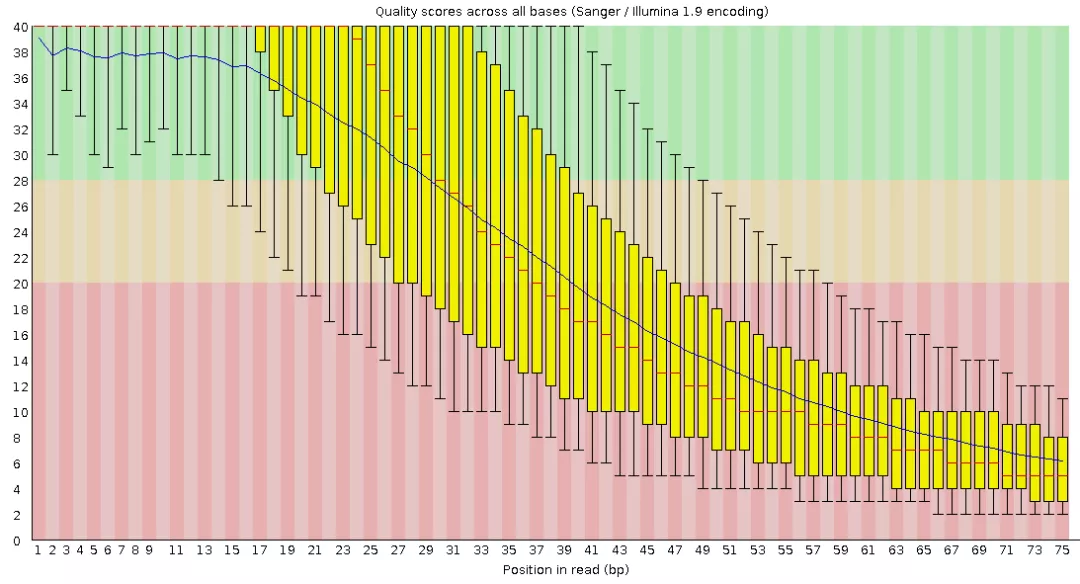
READ_QC/ERR011347/post-QC_report/final_pure_reads_2_fastqc.html包含质控前的质量评估结果。
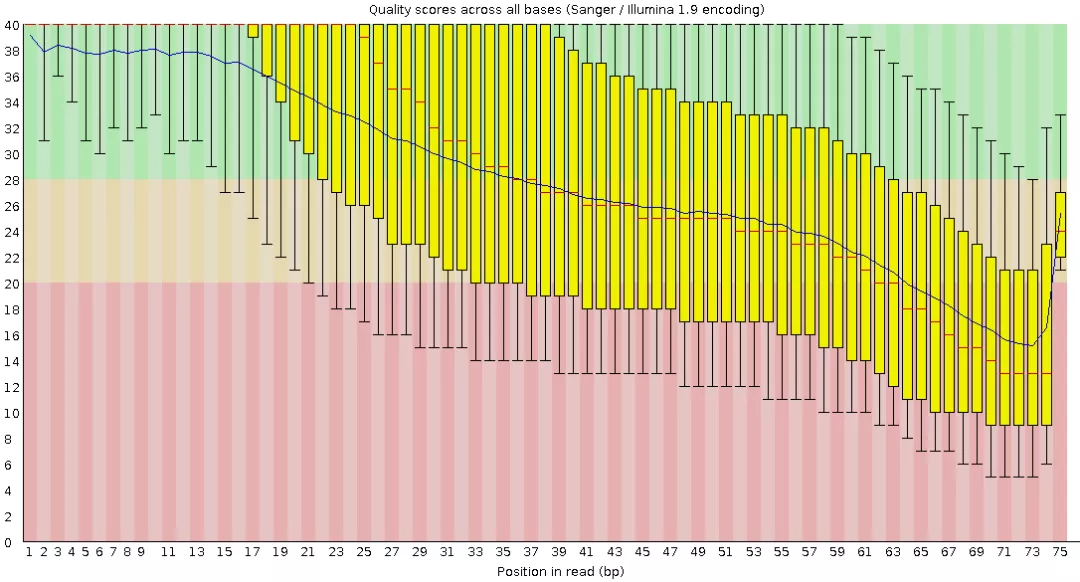
如果确定质量合格(质量不符合你自己要求,可查看帮助调整参数重新质控),可移动结果到新目录,质控后15G变为9G。
mkdir CLEAN_READS
for i in READ_QC/*; do
b=${i#*/}
mv ${i}/final_pure_reads_1.fastq CLEAN_READS/${b}_1.fastq
mv ${i}/final_pure_reads_2.fastq CLEAN_READS/${b}_2.fastq
done
2.2 组装
文件合并,方便拼接简化输入文件、组装获得统一的参考contig,如果文件过大,也可能需要分批处理。
cat CLEAN_READS/ERR*_1.fastq > CLEAN_READS/ALL_READS_1.fastq
cat CLEAN_READS/ERR*_2.fastq > CLEAN_READS/ALL_READS_2.fastq
组装,获得叠连群(contigs),是Binning的基本单元
#运行1h,仍然报错,去掉–metaspades后8m完成
time metawrap assembly -1 CLEAN_READS/ALL_READS_1.fastq -2 CLEAN_READS/ALL_READS_2.fastq -m 200 -t 96 -o ASSEMBLY # --metaspades
组装结果为ASSEMBLY/final_assembly.fasta,统计报告见ASSEMBLY/assembly_report.html
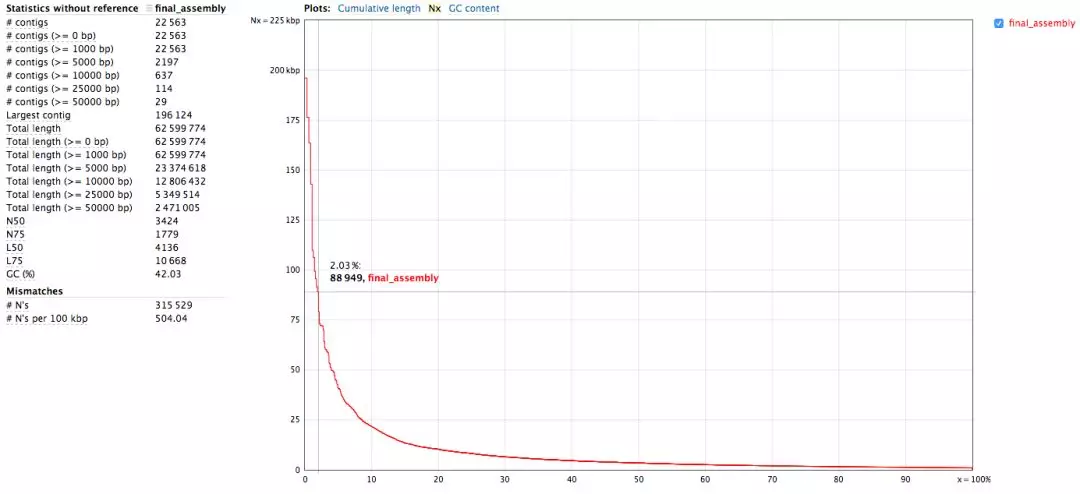
查看最长的3个contig信息
grep ">" ASSEMBLY/final_assembly.fasta | head -n3
>NODE_1_length_196124_cov_2.427049
>NODE_2_length_176373_cov_3.889994
>NODE_3_length_163601_cov_3.070200
看到最长的contig将近200kb,Top 3大于150kb,我们只使用了测试的7G数据,通常使用30 - 100 GB,拼接结果会更好。
参数说明:
Usage: metaWRAP assembly [options] -1 reads_1.fastq -2 reads_2.fastq -o output_dir
Options:
-1 STR forward fastq reads
-2 STR reverse fastq reads
-o STR output directory
-m INT memory in GB (default=24)
-t INT number of threads (defualt=1)
-l INT minimum length of assembled contigs (default=1000)
--megahit assemble with megahit (default)
--metaspades assemble with metaspades instead of megahit (better results but slower amd higher memory requirement)
2.3 kraken物种注释(可选)
kraken在reads和contig层面进行物种注释。当然kraken这么全能的工具,还可以在基因和bin层面
# 7G演示数据,8线程耗时3m17s
metawrap kraken -o KRAKEN -t 96 -s 1000000 CLEAN_READS/ERR*fastq ASSEMBLY/final_assembly.fasta
结果文件夹中有注释结果文件.kraken(每个reads的注释结果)、.krona(注释结果分类汇总),和可视化的Krona网页图表kronagram.html
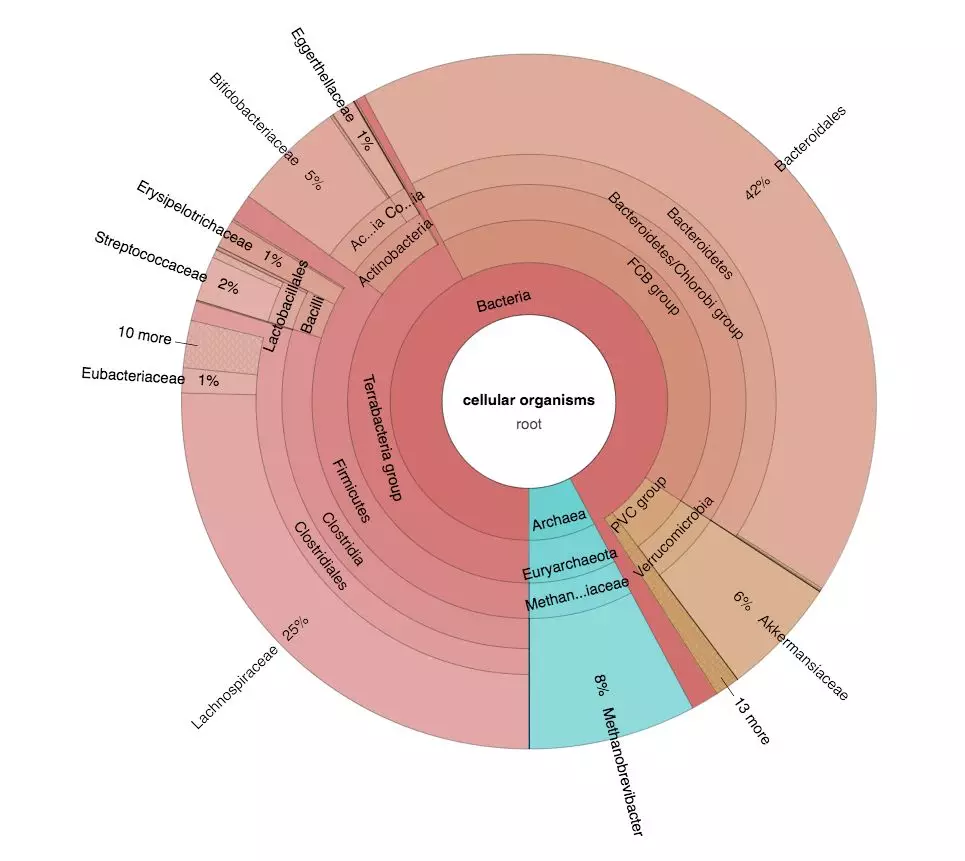
参数说明:
(metawrap-env) [sam@localhost 21]$ metawrap kraken --help
Run on any number of fasta assembly files and/or or paired-end reads.
Usage: metaWRAP kraken [options] -o output_dir assembly.fasta reads_1.fastq reads_2.fastq ...
Options:
-o STR output directory
-t INT number of threads
-s INT read subsampling number (default=all)
--no-preload do not pre-load the kraken DB into memory (slower, but lower memory requirement)
Note: you may pass any number of sequence files with the following extensions:
*.fa *.fasta (assumed to be assembly files) or *_1.fastq and *_2.fastq (assumed to be paired)
2.4 4. 三种方法Bin
基于contig和三种bin方法,需要拼接结果和原始序列,用时34m
metawrap binning -o INITIAL_BINNING -t 8 -a ASSEMBLY/final_assembly.fasta --metabat2 --maxbin2 --concoct CLEAN_READS/ERR*fastq
结果目录中文件或目录说明
insert_sizes.txt 样本量统计和估计的建库时文库片段大小
concoct_bins maxbin2_bins metabat2_bins 三个目录为三种bin的结果
work_files有三种bin分析所需要的文件,如不同格式的bin覆盖度或丰度信息。
concoct, metabat2和maxbin2三个软件分别获得了47, 29 和20个bins,但我们并不知道它们质量如何,可以添加–run-checkm参数获得质量评估
这一步输入序列的后缀名需要为 fastq, 如果为fq的话,都会报错
(metawrap-env) [sam@localhost 21]$ metawrap binning --help
metawrap binning --help
Usage: metaWRAP binning [options] -a assembly.fa -o output_dir readsA_1.fastq readsA_2.fastq ... [readsX_1.fastq readsX_2.fastq]
Note1: Make sure to provide all your separately replicate read files, not the joined file.
Note2: You may provide single end or interleaved reads as well with the use of the correct option
Note3: If the output already has the .bam alignments files from previous runs, the module will skip re-aligning the reads
Options:
-a STR metagenomic assembly file
-o STR output directory
-t INT number of threads (default=1)
-m INT amount of RAM available (default=4)
-l INT minimum contig length to bin (default=1000bp). Note: metaBAT will default to 1500bp minimum
--metabat2 bin contigs with metaBAT2
--metabat1 bin contigs with the original metaBAT
--maxbin2 bin contigs with MaxBin2
--concoct bin contigs with CONCOCT
--universal use universal marker genes instead of bacterial markers in MaxBin2 (improves Archaea binning)
--run-checkm immediately run CheckM on the bin results (requires 40GB+ of memory)
--single-end non-paired reads mode (provide *.fastq files)
--interleaved the input read files contain interleaved paired-end reads
2.5 Bin提纯
三种主流bin结果各有优缺点,我们将对结果进行整合和优化,以获得更好的结果
如果你有超过3种结果,也可以合并,如5种结果,先合并1,2,3;再合并4,5;最后将两类合并。
推荐使用完整度70,污染率5的阈值;但本演示测序数据较小,仅使用50和10级别的阈值,保证有足够的结果用于演示和评估。要求越高,bin越少,请根据个人需要调整。
主要参数有-o输出目录;-t线程数;-A/B/C三种Bin结果;-c完整度阈值;-x污染率阈值;如下8线程,耗时67m
metawrap bin_refinement -o BIN_REFINEMENT -t 8 -A INITIAL_BINNING/metabat2_bins/ -B INITIAL_BINNING/maxbin2_bins/ -C INITIAL_BINNING/concoct_bins/ -c 50 -x 10
结果目录中有三个原始bin的结果与统计。metaWRAP目录包含最终结果。如果想查看中间文件见Binning_refiner目录。
.stat文件包含每个bin的统计:完整性、污染率、GC含量、物种、N50、大小和来源。
如cat BIN_REFINEMENT/metaWRAP_bins.stats
bin completeness contamination GC lineage N50 size binner
bin.1 98.92 0.866 0.401 Clostridiales 13297 2373299 binsBC
bin.8 93.73 0.884 0.312 Euryarchaeota 5058 1485694 binsC
bin.2 85.57 0.223 0.370 Clostridiales 5890 2030996 binsA
提纯的结果如何看,详见BIN_REFINEMENT/figures/目录中的图片,有eps矢量图和Png位图供查看和修改发表使用,作者太贴心了!
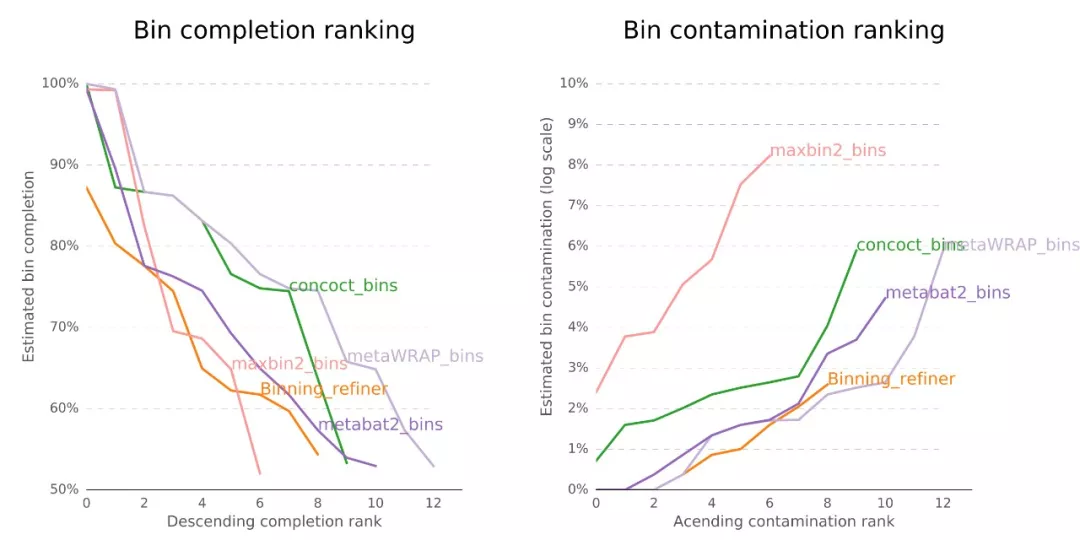
2.6 Blobology可视化bin
我们使用Blobology模块,将Contig的GC含量和丰度进行散点图可视化,并按物种或Bin进行着色,来观察结果。
可视化Bin的contig丰度和GC含量,耗时31m
metawrap blobology -a ASSEMBLY/final_assembly.fasta -t 8 -o BLOBOLOGY --bins BIN_REFINEMENT/metaWRAP_bins CLEAN_READS/ERR*fastq
存在如下图所 需的原始数据.binned.blobplot.文件,包括GC含量、丰度、平均丰度等,可使用ggplot2方便可视化如下图,难点是颜色分配。
参考脚本见程序目中
/conda2/envs/metawrap/bin/metawrap-scripts/blobology/makeblobplot_with_bins.R final_assembly.binned.blobplot 1 taxlevel_phylum有三个参数,脚本较老,需要自己修改一下。
按门水平着色的GC含量vs丰度散点图

按Bin着色的GC含量vs丰度散点图
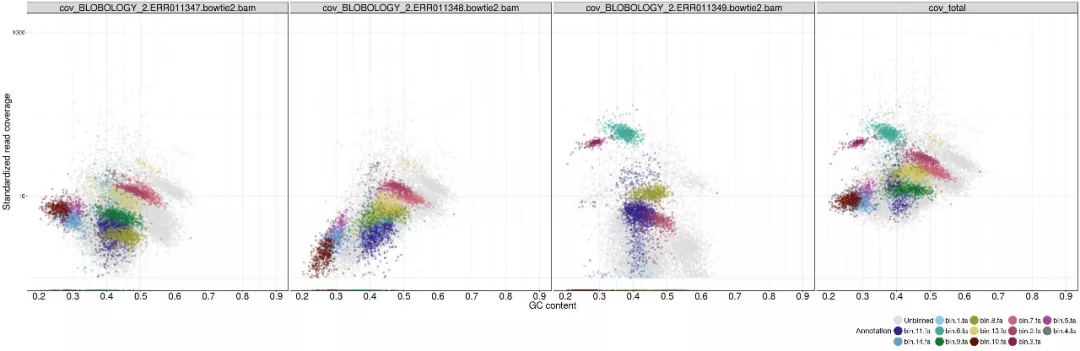
7. Bin定量
我们想知道这些单菌基因组草图(bin)在每个样品中的丰度。quant_bin模块帮你实现,它也依赖salmon来实现定量,估计每一个scaffold的丰度,以及bin的平均丰度。
实际时间1m39s,计算时间45m6s,使用调用了我的所有线程。此处可设置线程,但salmon仍尽可能多调用资源。
metawrap quant_bins -b BIN_REFINEMENT/metaWRAP_bins -t 8 -o QUANT_BINS -a ASSEMBLY/final_assembly.fasta CLEAN_READS/ERR*fastq
结果包括bin丰度热图QUANT_BINS/genome_abundance_heatmap.png。
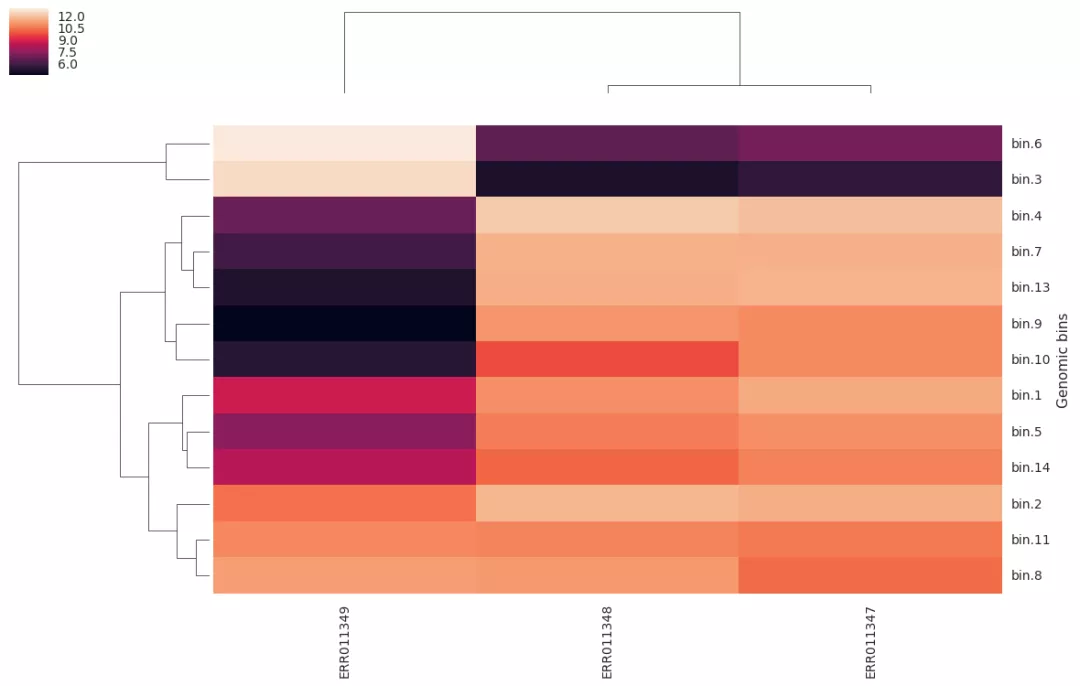
如果想自己画图,原始数据位于QUANT_BINS/abundance_table.tab
Genomic bins ERR011349 ERR011348 ERR011347
bin.9 0.113912116828 35.851964987 39.8440491514
bin.10 0.273774684856 9.52869077293 39.988244574
参数说明:
(metawrap-env) [sam@localhost 20200819]$ metawrap quant_bins
metawrap quant_bins
------------------------------------------------------------------------------------------------------------------------
----- Non-optional parameters -b and -o were not entered -----
------------------------------------------------------------------------------------------------------------------------
Usage: metaWRAP quant_bins [options] -b bins_folder -o output_dir -a assembly.fa readsA_1.fastq readsA_2.fastq ... [readsX_1.fastq readsX_2.fastq]
Options:
-b STR folder containing draft genomes (bins) in fasta format
-o STR output directory
-a STR fasta file with entire metagenomic assembly (strongly recommended!)
-t INT number of threads
2.8 重组装
提纯的bin还可以通过再组装进一步改善结果。基于原始reads对结果优化,只有结果更优的情况,才对结果进行更新。
先调用bwa比对原始序列到bin,使用SPAdes不同参数下(宽松和严谨)精细组装,去除小于1500bp的叠连群,checkM评估,结果统计,绘图。
用时41m, 线程总用时178m
metawrap reassemble_bins -o BIN_REASSEMBLY -1 CLEAN_READS/ALL_READS_1.fastq -2 CLEAN_READS/ALL_READS_2.fastq -t 8 -m 800 -c 50 -x 10 -b BIN_REFINEMENT/metaWRAP_bins
结果统计见BIN_REASSEMBLY/reassembled_bins.stats,发现3个bin通过严格模式下得到优化,6个通过宽松模式得到改进,4个没有变化。
bin completeness contamination GC lineage N50 size binner
bin.10.orig 65.78 0.0 0.263 Bacteria 3045 1159966 NA
bin.7.strict 54.94 0.671 0.501 Clostridiales 3947 1474089 NA
bin.4.permissive 99.32 1.342 0.408 Clostridiales 72135 2088821 NA
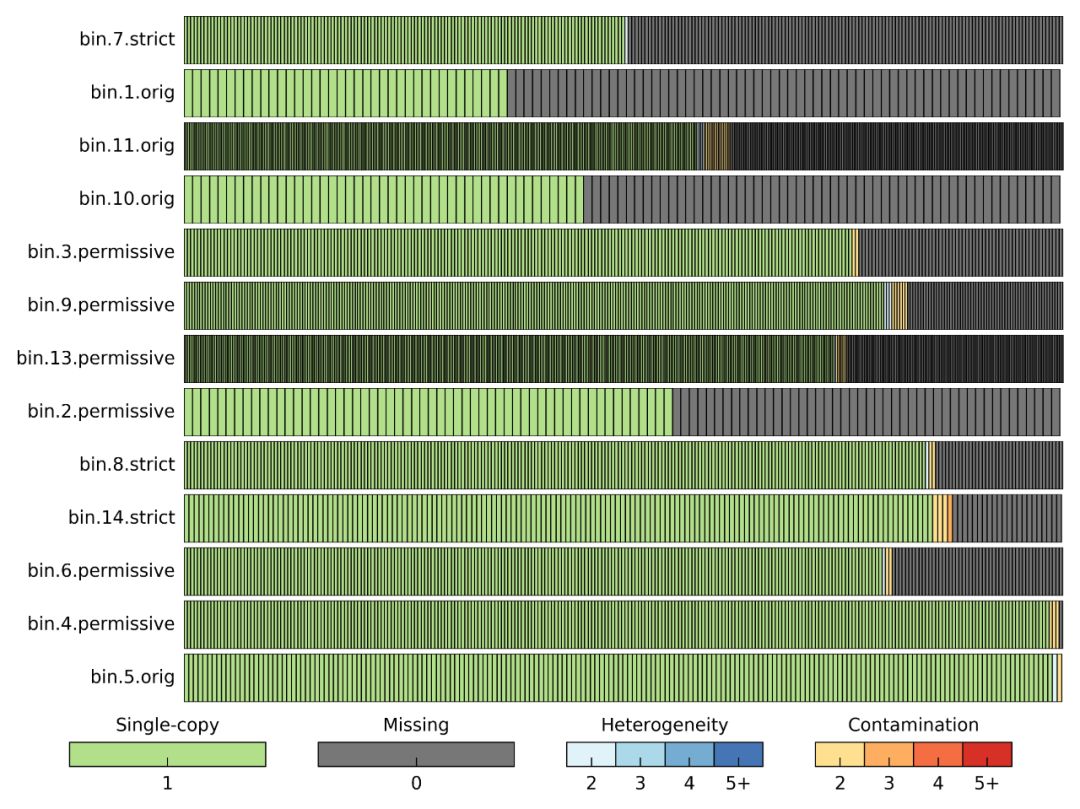
改进的情况如何呢?要查看结果BIN_REASSEMBLY/reassembly_results.png,比对重组装前后的变化,N50、这完整度和污染率均有改进。
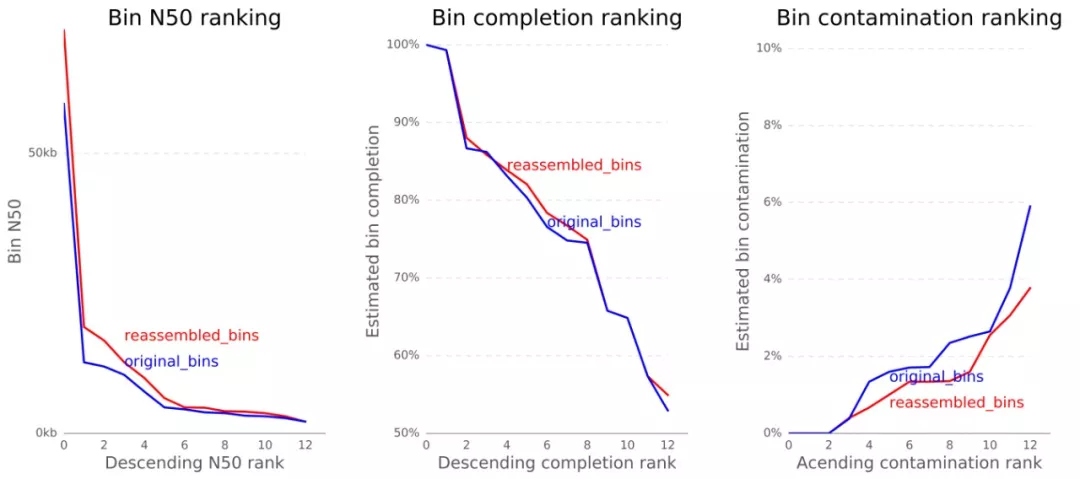
BIN_REASSEMBLY/reassembled_bins.png展示CheckM对bin评估结果的可视化。
2.9 bin物种注释
其实Bin提纯和重组装中,在checkM的stat文件中,就有物种的注释结果。但软件和数据库都不完善。
基于NCBI_nt和NCBI_tax数据库,我们使用 Taxator-tk 进行每条contig物种注释,再估计bin整体的物种。注释结果的准确性由参考数据库决定(如参考库中有错误,我们无法识别,只能将错就错)。
此步8线程用时12分钟。
# real 12m, user 84m
metawrap classify_bins -b BIN_REASSEMBLY/reassembled_bins -o BIN_CLASSIFICATION -t 8
注释结果见BIN_CLASSIFICATION/bin_taxonomy.tab
bin.1.orig.fa Bacteria;Firmicutes;Clostridia;Clostridiales
bin.5.orig.fa Archaea;Euryarchaeota;Methanobacteria;Methanobacteriales;Methanobacteriaceae;Methanobrevibacter;Methanobrevibacter smithii
bin.11.orig.fa Bacteria;Bacteroidetes;Bacteroidia;Bacteroidales;Bacteroidaceae;Bacteroides
注意物种注释层级不同,因为有些物种在数据库中没有近亲。
2.10 Bin功能注释
此步基于PROKKA进行基因注释。
# 4m5s
metaWRAP annotate_bins -o FUNCT_ANNOT -t 8 -b BIN_REASSEMBLY/reassembled_bins/
每个bin基因注释的gff文件见 FUNCT_ANNOT/bin_funct_annotations
head FUNCT_ANNOT/bin_funct_annotations/bin.1.orig.gff
NODE_75_length_31799_cov_0.983871 Prodigal:2.6 CDS 2866 3645 . - 0 ID=HMOHEJHL_00001;inference=ab initio prediction:Prodigal:2.6;locus_tag=HMOHEJHL_00001;product=hypothetical protein
NODE_75_length_31799_cov_0.983871 Prodigal:2.6 CDS 3642 4478 . - 0 ID=HMOHEJHL_00002;inference=ab initio prediction:Prodigal:2.6;locus_tag=HMOHEJHL_00002;product=hypothetical protein
NODE_75_length_31799_cov_0.983871 Prodigal:2.6 CDS 4606 5859 . - 0 ID=HMOHEJHL_00003;inference=ab initio prediction:Prodigal:2.6;locus_tag=HMOHEJHL_00003;product=hypothetical protein
基因蛋白和核酸序列见FUNCT_ANNOT/bin_translated_genes和 FUNCT_ANNOT/bin_untranslated_genes目录。PROKKA输出结果见 FUNCT_ANNOT/prokka_out.
五、报错
报错1:BLAST Database error: Error: Not a valid version 4 database.
(metawrap-env) [sam@g01 21]$ blastn -task megablast -num_threads 40 -db /data/database/metawrap/NCBI_nt/nt -outfmt '6 qseqid qstart qend qlen sseqid staxids sstart send bitscore evalue nident length' -query test.fasta
BLAST Database error: Error: Not a valid version 4 database.
解决办法:
blast的版本和数据库的版本不一致
wget -c ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-2.10.1+-x64-linux.tar.gz
tar xzvfm ncbi-blast-2.10.1+-x64-linux.tar.gz
/data/software/blast/ncbi-blast-2.10.1+/bin
export PATH=/data/software/blast/ncbi-blast-2.10.1+/bin:$PATH
参考资料
