【4.1】宏基因组分析--MetaWRAP
MetaWRAP这是一套强大的宏基因组分析流程,专注于宏基因组Binning。文章于2018年9月15日发表于《Microbiome》。文章简介见参考文献链接。
一、简介
- MetaWRAP是一款整合了质控、拼接、分箱、提纯、评估、物种注释、丰度估计、功能注释和可视化的分析流程,纳入超140个工具软件,可一键安装;
- 流程整合了CONCOCT、MaxBin、 metaBAT等三款分箱工具以及提纯和重组装算法;
- 与以上三种工具单独使用,以及与使用DAS_tool、Binning-refiner相比,分箱结果更佳。
- 在此基础上,MetaWRAP还可实现宏基因组分析从原始数据到结果可视化的全部流程,同时也可灵活使用各个模块独立分析,弹性多变。
工作原理
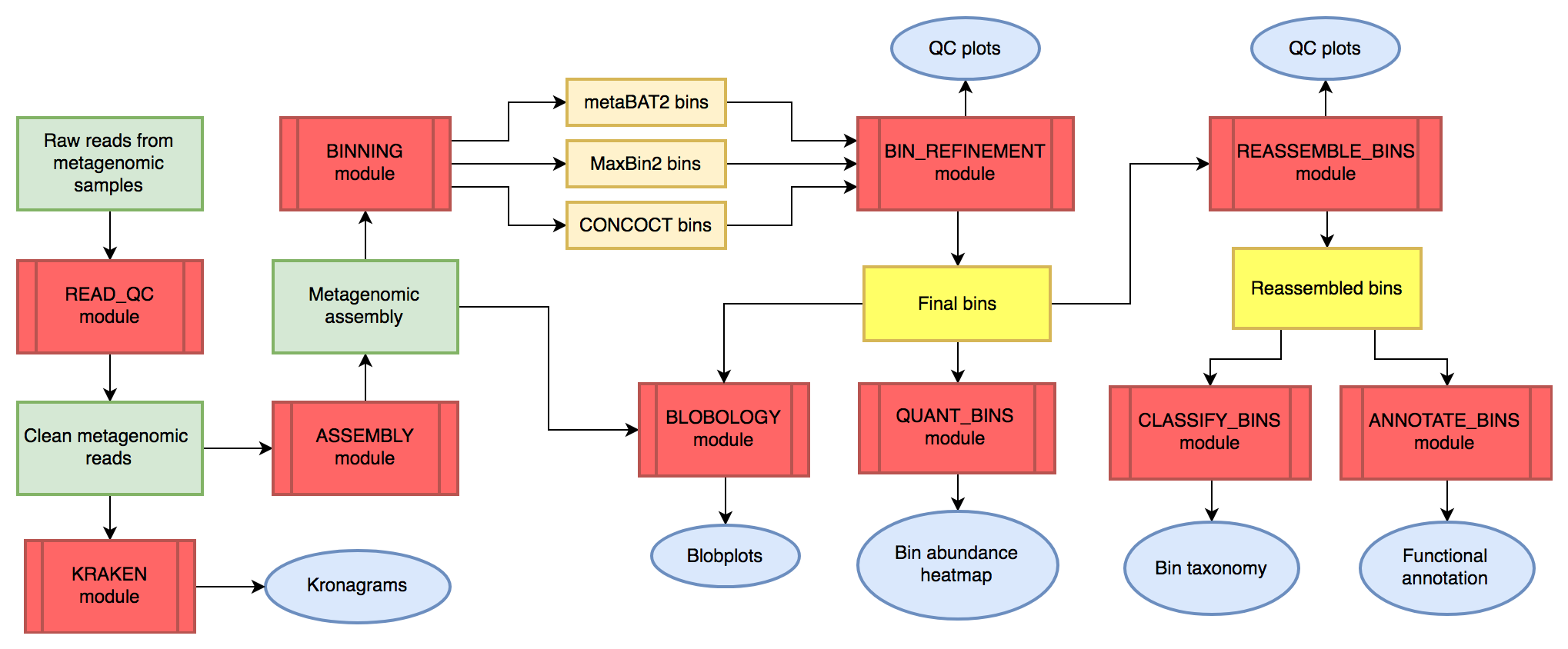
图中红色代表分析模块,绿色代表宏基因组数据,橙色代表中间文件,蓝色代表结果图表。
实现原始序列的质控、物种注释和可视化、宏基因组拼接、三种主流Bin方法分析和结果筛选与可视化、Bin的重新组装、Bin的物种和功能注释等。轻松实现Bin相关分析和可视化的绝大部分需求。
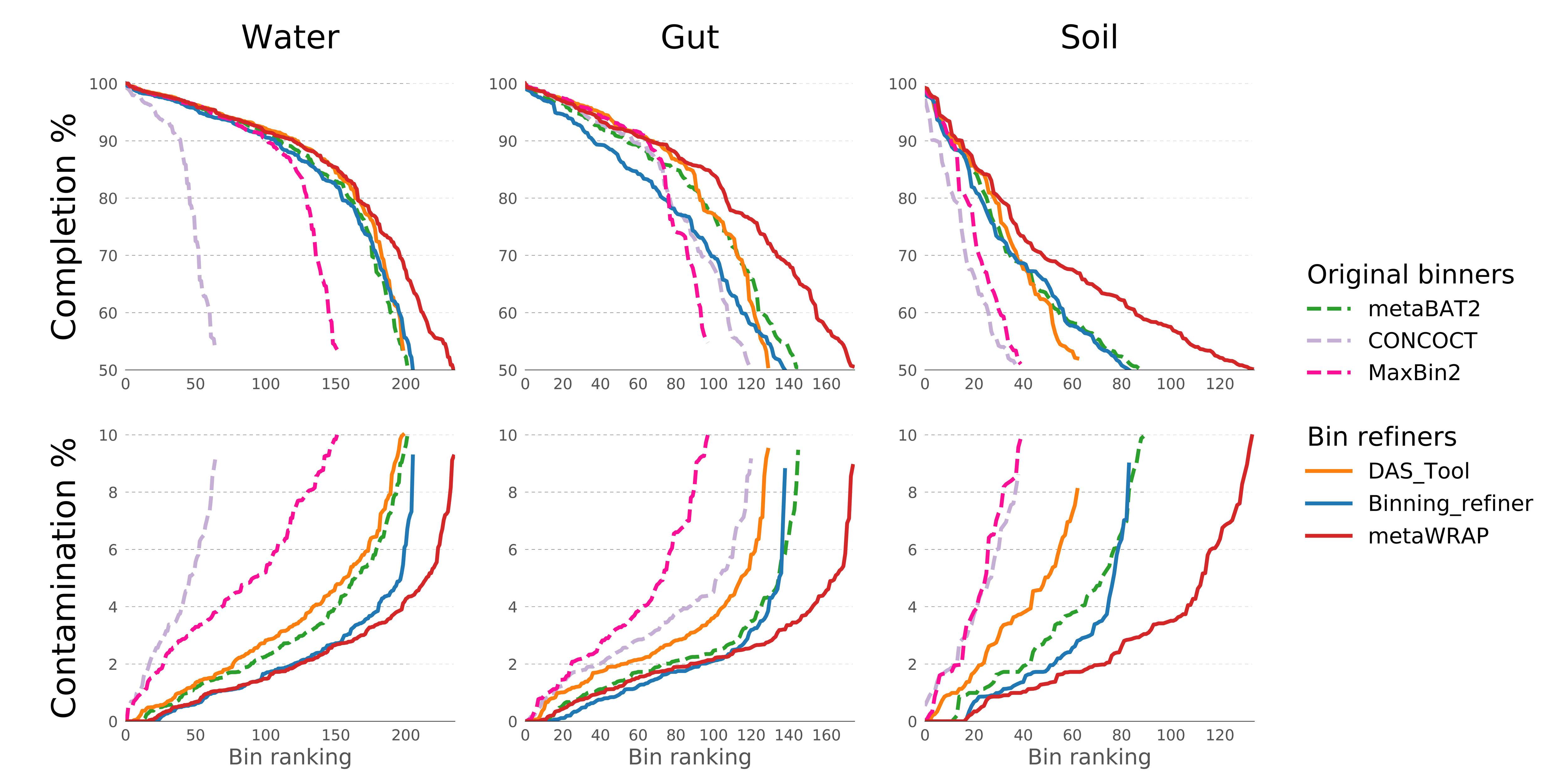
基于CAMI人工数据集高、中、低数据量下,对6款Bin软件结果的完整度和污染率进行评估。结果表明metaWRAP在各种情况下在完整度和污染率方面都表现更优秀。
功能模块
宏基因组数据预处理模块:
- 质控Read_QC: read质控剪切和移除人类宿主
- 组装Assembly: 质控、使用megahit或metaSPAdes拼接
- 物种注释Kraken: 对reads和contigs层面进行可视化
- 分箱Bin处理模块
分箱Binning:
- 利用MaxBin2, metaBAT2, 和CONCOCT三个软件分别分箱;
- 提纯Bin_refinement:对多种Bin结果评估和综合分析,获得更好的结果;
- 重组装Reassemble_bins:利用原始序列和评估软件二次组装,改善Bin的N50、完整度4) 定量Quant_bins: 估计样品中每个bin的丰度并热图展示
- 气泡图Blobology: blobplots可视化群体的contigs的物种和Bin分布
- 物种注释Classify_bins: 对Bin物种注释
- 基因注释Annotate_bins: 预测Bin中的基因
二、安装
开源代码:https://github.com/bxlab/metaWRAP
2.1 系统要求:
系统要求是由处理的数据量决定的。
-
其中一些软件,如KRAKEN、metaSPAdes对内存需求较高,推荐服务器至少8+核,64+GB内存,仅支持64位Linux系统。
-
对于300 GB以上数据用户,推荐配置48核,512内存或更高。
-
软件原作者的教程中参数使用了96线程和900G内存,可以推断软件开发和测试所用服务器至少为96线程和1TB内存。
2.2 安装软件
通过安装conda2
安装conda
wget https://repo.continuum.io/miniconda/Miniconda2-latest-Linux-x86_64.sh
bash Miniconda2-latest-Linux-x86_64.sh
#conda 选择安装在 /usr/local/miniconda2
启动conda,因为我默认的conda环境是conda3,所以这里一定要启动conda2
source /usr/local/miniconda2/bin/activate root
简化conda启动
vim ~/.bash_profile
alias activate2="source /usr/local/miniconda2/bin/activate root"
以后activate2后,就启动conda2的环境
安装metawrap
activate2
conda create -y -n metawrap-env python=2.7
conda activate metawrap-env
# Note: ordering is important
conda config --add channels defaults
conda config --add channels conda-forge
conda config --add channels bioconda
conda config --add channels ursky
conda install -y -c ursky metawrap-mg
# To fix the CONCOCT endless warning messages in metaWRAP=1.2+, run
conda install -y blas=2.5=mkl
# OR
conda install biopython blas=2.5 blast=2.6.0 bmtagger bowtie2 bwa checkm-genome fastqc kraken=1.1 krona=2.7 matplotlib maxbin2 megahit metabat2 pandas prokka quast r-ggplot2 r-recommended salmon samtools=1.9 seaborn spades trim-galore
# Note: this last solution is universal, but you may need to manually install concoct=1.0 and pplacer.
安装完以后会提示:
Krona installed. You still need to manually update the taxonomy
databases before Krona can generate taxonomic reports. The update
script is ktUpdateTaxonomy.sh. The default location for storing
taxonomic databases is /usr/local/miniconda2/envs/metawrap-env/opt/krona/taxonomy
If you would like the taxonomic data stored elsewhere, simply replace
this directory with a symlink. For example:
rm -rf /usr/local/miniconda2/envs/metawrap-env/opt/krona/taxonomy
mkdir /path/on/big/disk/taxonomy
ln -s /path/on/big/disk/taxonomy /usr/local/miniconda2/envs/metawrap-env/opt/krona/taxonomy
ktUpdateTaxonomy.sh
\ The default QUAST package does not include:
* GRIDSS (needed for structural variants detection)
* SILVA 16S rRNA database (needed for reference genome detection in metagenomic datasets)
* BUSCO tools and databases (needed for searching BUSCO genes) -- works in Linux only!
To be able to use those, please run
quast-download-gridss
quast-download-silva
quast-download-busco
三、 配置数据库
主要大小和依赖模块如下:
| Database | Size | Used in module |
|---|---|---|
| Checkm | 1.4GB | binning, bin_refinement, reassemble_bins |
| KRAKEN | 192GB | kraken |
| NCBI_nt | 99GB | blobology, classify_bins |
| NCBI_tax | 283MB | blobology, classify_bins |
| Indexed hg38 | 34GB | read_qc |
可以看到配置文件路径,一会需要修改这个文件
which config-metawrap
/usr/local/miniconda2/envs/metawrap-env/bin/config-metawrap
2.3.1 CheckM数据库 (ok)
下载文件276MB,解压后1.4GB
cd /home/sam/database
mkdir checkm
cd checkm
wget -c https://data.ace.uq.edu.au/public/CheckM_databases/checkm_data_2015_01_16.tar.gz
tar -xvf *.tar.gz
rm *.gz
checkm data setRoot
这个时候输入路径:/home/sam/database/checkm
2.3.2 KRAKEN数据库
下载建索引需要 > 300GB以上空间,完成后占用192GB空间
cd /home/sam/database
mkdir kraken
kraken-build --standard --threads 24 --db kraken --use-wget
kraken-build --db kraken --clean
vim /usr/local/miniconda2/envs/metawrap-env/bin/config-metawrap
KRAKEN_DB=/path/to/my/database/MY_KRAKEN_DATABASE
因为网速原因,一直下载不了,所以自己手动下载
(metawrap-env) [sam@localhost bin]$ which kraken-build
/usr/local/miniconda2/envs/metawrap-env/bin/kraken-build
下面代码注释掉:
#sub download_taxonomy {
# exec "download_taxonomy.sh";
#}
将这里面的代码:
/usr/local/miniconda2/envs/metawrap-env/libexec/download_taxonomy.sh
手动下载:
cd /home/sam/database/kraken/taxonomy
ftp://ftp.ncbi.nlm.nih.gov/pub/taxonomy/accession2taxid/nucl_gb.accession2taxid.gz
ftp://ftp.ncbi.nlm.nih.gov/pub/taxonomy/accession2taxid/nucl_wgs.accession2taxid.gz
ftp://ftp.ncbi.nlm.nih.gov/pub/taxonomy/taxdump.tar.gz
还有这两个
touch accmap.dlflag
touch taxdump.dlflag
gunzip *accession2taxid.gz
tar zxf taxdump.tar.gz
vim /usr/local/miniconda2/envs/metawrap-env/bin/config-metawrap
KRAKEN_DB=/home/sam/database/kraken
2.3.3 NCBI_nt
41GB,我下载大约12h;解压后99GB
cd /home/sam/database
mkdir NCBI_nt && cd NCBI_nt
wget -c "ftp://ftp.ncbi.nlm.nih.gov/blast/db/nt.*.tar.gz"
for a in nt.*.tar.gz; do tar xzf $a; done
vim /usr/local/miniconda2/envs/metawrap-env/bin/config-metawrap
BLASTDB=/home/sam/database/NCBI_nt
2.3.4 NCBI物种信息 (ok)
压缩文件45M,解压后351M
cd /home/sam/database
mkdir NCBI_tax ; cd NCBI_tax
wget -c ftp://ftp.ncbi.nlm.nih.gov/pub/taxonomy/taxdump.tar.gz
tar -xvf taxdump.tar.gz
vim /usr/local/miniconda2/envs/metawrap-env/bin/config-metawrap
TAXDUMP=/home/sam/database/NCBI_tax
2.3.5 人类基因组bmt索引 (ok)
下载人类基因组942M,解压后合并3.2G,并建索引34GB
cd /home/sam/database
mkdir BMTAGGER_INDEX ; cd BMTAGGER_INDEX
wget -c ftp://hgdownload.soe.ucsc.edu/goldenPath/hg38/chromosomes/*fa.gz
gunzip *fa.gz
cat *fa > hg38.fa
rm chr*.fa
bmtool -d hg38.fa -o hg38.bitmask
srprism mkindex -i hg38.fa -o hg38.srprism -M 10000
vim /usr/local/miniconda2/envs/metawrap-env/bin/config-metawrap
添加环境变量:
BMTAGGER_DB=/home/sam/database/BMTAGGER_INDEX
五、报错:
报错1
(metawrap-env) [sam@localhost BMTAGGER_INDEX]$ srprism mkindex -i hg38.fa -o hg38.srprism -M 100000
memmgr.cpp:84 [2] allocation error (request: 103251411536 bytes)
ERROR: memmgr.cpp:84 [2] allocation error (request: 103251411536 bytes) <srprism.cpp:850>
解决办法:
-M 后面接的数值调小,例如:
报错2
提示找不到perl的库
export PERL5LIB=/data/user/sam/project/meta/miniconda2/envs/metawrap-env/lib/5.26.2
参考资料
