【2.3.3】肿瘤等位基因特异性拷贝数分析(ASCAT)
- github: https://github.com/VanLoo-lab/ascat
- https://www.crick.ac.uk/research/labs/peter-van-loo/software
This repository provides the ASCAT R package (v3.1.2) that can be used to infer tumour purity, ploidy and allele-specific copy number profiles.
ASCAT is described in detail in: Allele-specific copy number analysis of tumors. Van Loo P et al. PNAS (2010).
This repository also contains the code underlying additional publication: Allele-specific multi-sample copy number segmentation. Ross EM, Haase K, Van Loo P & Markowetz F. Bioinformatics (2020).
更多介绍:https://www.crick.ac.uk/research/labs/peter-van-loo/software
一、研究背景
ASCAT is a method to derive copy number profiles of tumor cells, accounting for normal cell admixture and tumor aneuploidy. ASCAT infers tumor purity (the fraction of tumor cells) and ploidy (the amount of DNA per tumor cell, expressed as multiples of haploid genomes) from SNP array or massively parallel sequencing data, and calculates whole-genome allele-specific copy number profiles (the number of copies of both parental alleles for all SNP loci across the genome).
与正常基因组相比,癌症基因组可发生单碱基突变到染色体片段的插入与缺失,乃至整个基因组复制等变化。CGH、SNP芯片和全基因组测序等方法被广泛用于绘制癌症基因组。但是肿瘤细胞染色体的改变使得难以构建和解释上述方法得到的数据。染色体所有基因座拷贝数的识别计算是正确解释癌症基因组的必要部分。
ASCAT(Allele-specificcopy number analysis of tumors):可用于肿瘤基因座等位基因特异拷贝数的分析。该方法综合考虑了肿瘤样本中的异常细胞和非异常细胞、肿瘤的倍性,可以计算异常细胞的百分比、肿瘤倍性、扩增、缺失、LOH、等位基因的偏度。基于肿瘤基因表达谱芯片和SNP芯片的整合分析,可以计算差异表达基因表达水平变化、以及在基因组的拷贝数变化。同时可得到转录因子、癌基因、原癌基因的等位基因的拷贝数变化,并研究基因的LOH。基因表达水平和相关等位基因的拷贝数变化可帮助我们进一步分析肿瘤样本与正常对照组的差异。
二、分析流程展示
该流程的数据类型为肿瘤基因表达谱芯片(AFFY平台和Illumina平台的芯片均可),以及相对应的SNP芯片(目前为llumina平台的芯片)
生物信息学分析步骤包括:
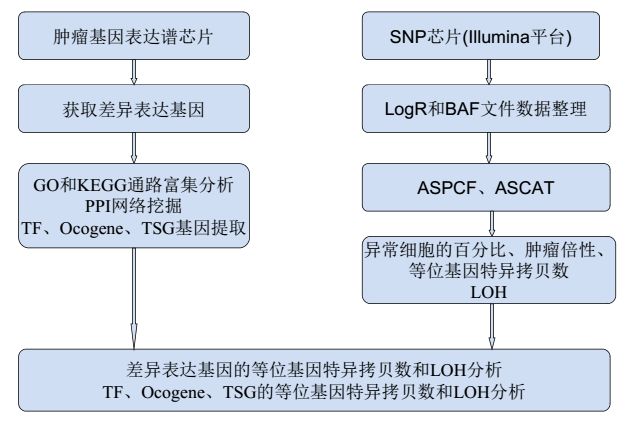
三、示例结果
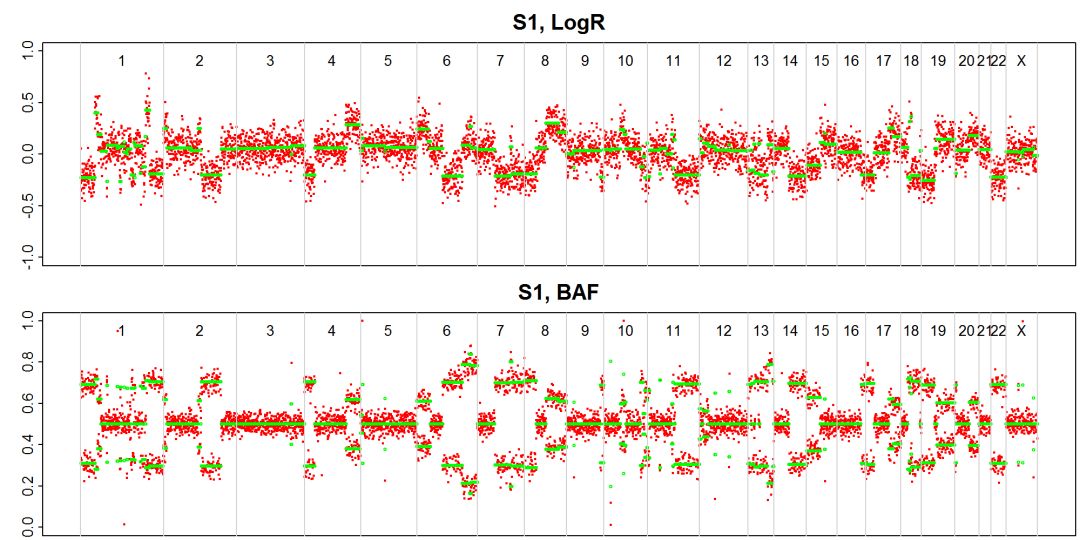
图1 乳腺癌LogR和BAF数据的预处理

图2 乳腺癌样本中Aberrant franction和Ploidy的预测
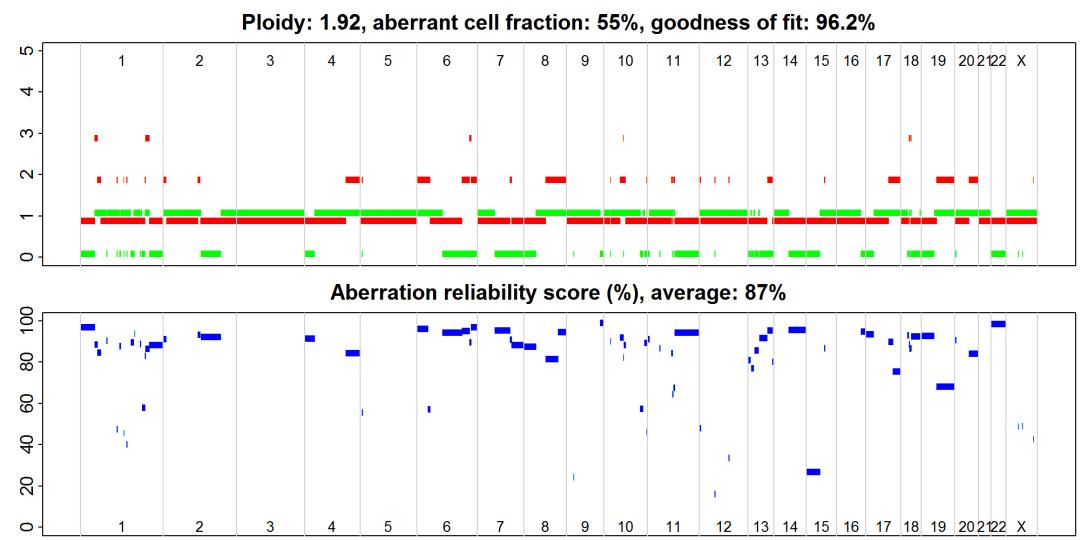
图3 乳腺癌allele-specificcopy number profiling
四、讨论
4.1 我可以使用ASCAT进行(种系)CNV分析吗?
ASCAT是检测癌症样本中体细胞拷贝数改变(CNA,somatic copy number alterations )的工具,不能用于检测种系拷贝数变异(CNVs,germline copy number variants)。术语CNV是指一种种种系变异,在群体中具有多态性。为了避免混淆,对于肿瘤样本中体细胞拷贝数的变化,我们建议始终使用术语CNA。
4.2 ASCAT也可以应用于细胞系吗?
ASCAT将在匹配的细胞系数据上工作良好。然而,它不太适合分析不匹配的细胞系数据,因为种系基因型预测工具利用来自混合的正常细胞的信号来推断种系基因。由于大多数细胞系在实践中是不匹配的,ASCAT很可能不是分析细胞系数据的理想方法。
4.3 我什么时候应该使用ASCAT?我什么时候应该使用Battenberg?
Battenberg算法是专门为检测全基因组测序数据上的亚克隆拷贝数变化而设计的。目前版本的Battenberg也可以从数据中推断纯度和倍性,这将是我们分析全基因组测序数据的首选方法。
对于其他测序数据(外显子组或靶向)的分析,单倍型分期(haplotype phasing )的附加值更有限,我们建议使用ASCAT。ASCAT还支持对其他物种的数据进行分析,并支持对广泛的SNP阵列进行分析。
参考资料
