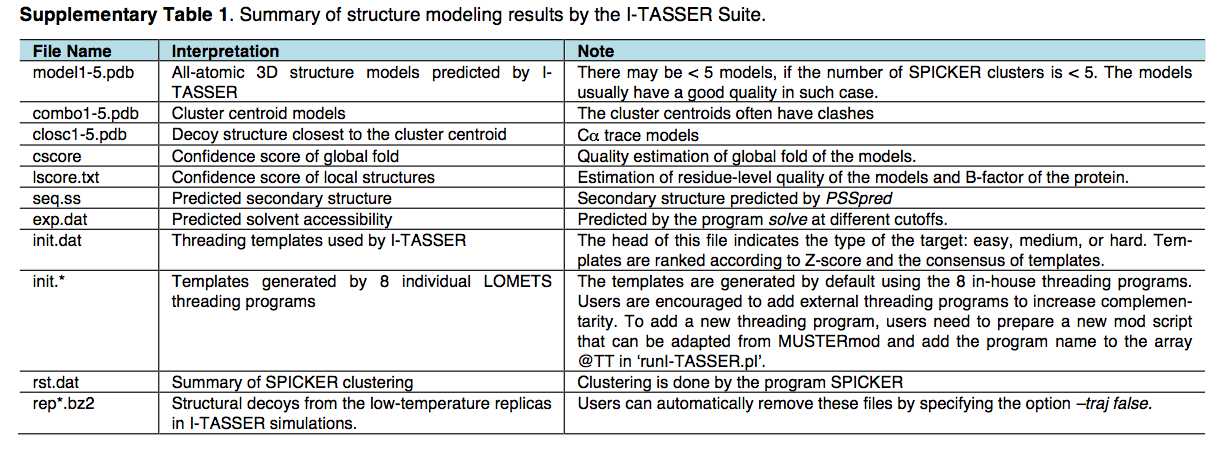
【4.5.1】蛋白质结构和功能预测(I-TASSER)
致编辑:所有生物体的所有基因和基因产物(例如蛋白质)的结构和功能分配,是这个后基因组时代的主要挑战。 在这里,我们介绍了I-TASSER套件( http://zhanglab.ccmb.med.umich.edu/I-TASSER/download/ ),它是用于蛋白质结构和功能建模的独立软件包。
具有已知序列的蛋白质的数量与具有实验表征的结构和功能的蛋白质的数量之间的差距不断增大。缩小这种差距的一种方法是开发先进的计算方法,以便从序列建模结构和功能,最近在社区范围的盲实验(community-wide blind)中已经看到了进展。
I-TASSER最初是设计用于通过迭代线程装配模拟( terative threading assembly simulations )进行蛋白质结构建模的。最近,通过将结构预测与已知功能模板进行匹配,将其扩展为基于结构的功能注释。在这里,我们介绍I-TASSER Suite,这是一个独立的程序包,用于实现基于I-TASSER的蛋白质结构和功能建模管道。尽管建立了在线I-TASSER服务器并在社区中广泛使用,但是来自单个实验室的有限计算资源阻止了这些算法的大规模应用。我们期望独立软件包的开发将消除计算资源的障碍,从而实现对新结构和功能建模方法的基准测试。
I-TASSER Suite流程包括四个常规步骤:
- 线程模板(threading template)识别,
- 迭代结构装配模拟,
- 模型选择和完善
- 基于结构的功能注释(图1a)。
第一步,LOMETS将查询通过非冗余结构库进行查询以识别结构模板。 LOMETS是一种包含八种折叠识别程序(PPAS,Env-PPAS,wPPAS,dPPAS,dPPAS2,wdPPAS,MUSTER和wMUSTER)的元线程(meta-threading )方法。这些程序通常基于序列图谱间的比对(sequence profile-to- profile alignments),但结合了各种结构特征(补充方法)。这种变化对于产生互补的比对是重要的,互补的比对增加了模板检测的覆盖范围。

图1 I-TASSER Suite用于蛋白质结构和功能建模的流程图和说明性示例。 (a)I-TASSER Suite管道的流程图,包括基于LOMETS的模板识别,基于I-TASSER的蒙特卡洛(MC)结构装配仿真和基于COACH的功能注释。 (b–g)蛋白质结构和功能预测的例子,其中I-TASSER和X射线衍射实验显示的蛋白质结构分别以蓝色和红色卡通形式表示,配体结合残基在卡通上用棒突出显示。通过COACH和实验得到的配体结构分别显示为黄色stick-and-ball forms and sticks。补充表3中列出了所有目标的建模参数。图中显示了假细菌拟杆菌(Bacteroides thetaiotaomicron)的假设蛋白BT_4147(CASP ID:R0006; PDB ID:4e0eA) (b)来自人的白细胞介素34(CASP:R0007; PDB:4dkcA)(c),来自大肠杆菌的镁和钴外排蛋白CorC与单磷酸腺苷结合(CASP:T0652; PDB:4hg0A)(d),洋葱伯克霍尔德菌的营养结合蛋白与蛋氨酸结合(CAMEO:C0081; PDB:4qhqA)(e),大肠杆菌的N-酰基转移酶与乙酰辅酶A结合(CAMEO:C0050; PDB:4qvtA)(f)和葡萄球菌的热核酸酶金黄色素与钙离子和3,5-二磷酸胸苷结合(CAMEO:C0046; PDB:4qf4A)(g)。 (h)T0652残基水平距离误差估计的示例,其中实线是预测误差,虚线是I-TASSER模型相对于实验结构的实际误差。底部,二级结构预测;红色箭头表示配体结合位点。
在查询到模板对齐之后,该序列分为threading-aligne区域和threading-unaligned 区域。全长模型的拓扑结构是通过重新组装从模板上切除的连续排列的片段而构建的,其中未排列区域的结构是通过从头开始折叠从头开始构建的。在优化的基于知识的力场的指导下,通过复制-交换蒙特卡罗模拟进行结构折叠和重新组装,该力场包括三个主要部分:(i)通用统计势能,(ii)氢键网络和(iii) LOMETS(补充方法)中基于线程的约束(threading-based restraints)。
最低的自由能构象通过结构聚类确定。从质心模型(centroid models)开始进行第二轮装配仿真,以消除空间冲突并优化全局拓扑。最终的原子结构模型是通过两步原子级的能量最小化方法从低能构象构建的。全局模型的正确性通过置信度得分进行评估,该置信度得分基于线程(threading)对齐的重要性和结构聚类的密度。通过新开发的方法ResQ评估结构模型的残基水平局部质量和目标蛋白的B因子,该方法建立在建模模拟的变化和通过支持向量回归训练的同源比对的不确定性的基础上。
对于功能注释,将具有最高置信度得分的结构模型与配体-蛋白质相互作用的BioLiP5数据库进行匹配,以检测同源功能模板。从功能模板中可以得出有关配体结合位点(LBS),酶委员会(EC)和基因本体论(GO)的功能见解。我们开发了三种互补算法(COFACTOR,TM-SITE和S-SITE)来增强功能推断,它们的共识是使用支持向量机由COACH4得出的。套件的安装,实施和结果解释的详细说明可以在补充方法和补充表1和2中找到。
I-TASSER Suite管道已在最近的社区范围内的结构和功能预测实验中进行了测试,包括CASP10(参考文献1)和CAMEO2。总体而言,对于CASP10中的36个“新折叠”(NF)目标中的10个,I-TASSER产生了正确的折叠,模板建模得分(TM得分)> 0.5,其中蛋白质数据库(PDB)中没有同源模板)。在110个基于模板的建模目标中,有92个的TM得分> 0.5,有89个在相同的螺纹对齐区域将模板拉近了本机,均方根偏差平均改善了1.05Å。在CAMEO中,COACH为4,271个目标生成LBS预测,平均准确度为0.86,比实验中第二好的方法高20%。
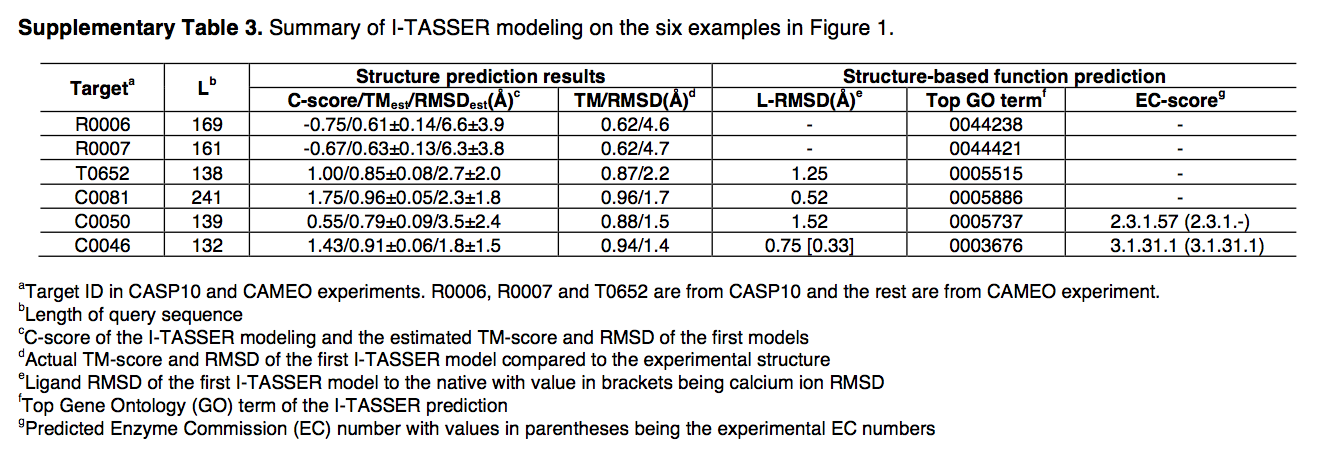
在这里,我们使用来自社区盲测1,2的六个示例(图1b–g)来说明基于I-TASSER Suite的结构和功能建模。 R0006和R0007是来自CASP10的两个NF目标,并且I-TASSER构建了正确折叠的模型,两个目标的TM得分均为0.62(图1b,c)。图中显示了针对T0652的ResQ进行的本地质量估算,与模型与本地模型的实际偏差相比,该模型的平均误差为0.75Å(图1h)。四个LBS预测实例(图1d–g)来自CASP10(参考文献1)和CAMEO2; COACH生成的配体模型均具有配体r.m.s.偏差低于2Å。 COACH还正确地将三位数和四位数的EC编号分配给了酶标靶C0050和C0046(补充表3)。
总之,我们开发了一个独立的I-TASSER Suite,可用于离线蛋白质结构和功能预测。 核心程序已经在基准测试和盲法实验中进行了广泛测试,证明了其相对于其他最新方法的优势。 此外,该套件还包含许多新开发功能,包括六种内部线程算法,用于局部质量和B因子估计的ResQ以及EC和GO预测算法; 这些发展对于使I-TASSER Suite成为有效的独立工具进行蛋白质结构和功能预测至关重要。 该软件包应使生物医学界能够进行大规模应用。
二、方法
2.1 Threading program
2.1.1 PPAS
PPAS是序列图-图比对方法(sequence profile–profile alignment ),辅以二级结构匹配。 PPAS的评分功能定义为
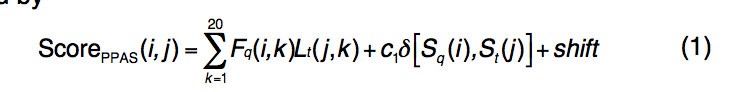
-
其中Fq(i,k)代表通过NCBI非冗余序列数据库通过PSI-BLAST搜索进行的3次迭代和E值截止= 0.001的MSA,第i个位置的第k个氨基酸的频率。 利用Henikoff加权来减少频率分布计算中对齐的多个序列的冗余。
-
Lt(j,k)表示在第j个位置的第k个氨基酸的模板的对数奇数图(log-odd),其通过PSI-BLAST搜索针对每个模板预先计算。
-
Sq(i)代表目标序列第i个位置的二级结构,
-
St(j)代表模板第j个位置的二级结构。
-
δ[Sq(i),St(j)]是Kronecker delta函数,如果 Sq(i)= St(j),则值为1,否则= 0。
-
查询的二级结构由PSSpred预测,而模板的二级结构由STRIDE分配,该结构包含三种状态:α-螺旋(H),β-链(E)和coil(C)。
-
引入shift参数以避免局部区域中无关残基的对齐。
Needleman-Wunsch动态规划算法用于搜索查询和模板序列之间的最佳匹配,而忽略结束空位罚分。使用一组300个非冗余训练蛋白优化了c1(= 0.65),shift(= -0.96),空位开放(go = -7.0)和空位延伸(ge = -0.54)的参数最大化线程模型(threading models)的平均TM得分6。
2.1.2 Env-PPAS
Env-PPAS的比对评分功能类似于PPAS,但增加了新的环境潜力(environment potential):
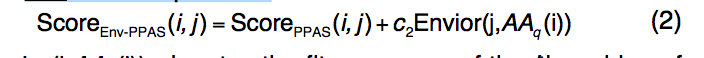
其中Envior(j,AAq(i))表示第i个残基的适拟合得分, 在模板结构的第j个残基的结构环境中查询AAq(i)。
这种潜力描述了残留物对特定骨架扭转角的倾向,溶剂可及性,二级结构和侧链取向的特定接触[7,8],这些接触是根据准化学近似法从5606个非冗余PDB结构的统计数据中得出的。 。 为了提高算法对随机比对的敏感性,请针对反向序列对比对分数进行重新归一化:

RScoreEnv-PPAS是Env-PPAS在人工序列上的原始比对得分,该序列构成查询序列的逆转,L表示查询序列的长度。 c2(= 0.45)是根据我们的训练数据集确定的。 Smith-Waterman局部动态编程算法用于识别查询和模板之间的最佳对齐方式。
2.1.3 wPPAS
wPPAS是PPAS的扩展,它引入了一个新的加权序列配置文件,该配置文件由不同的加权方案构成。
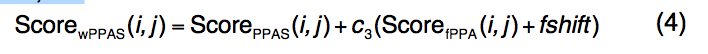
ScorefPPA(i,j)的评分函数定义为
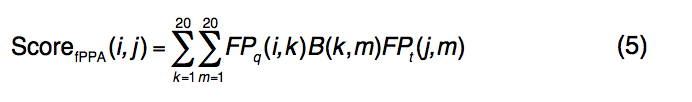
在等式中 (5),
- FPq(i,k)表示查询序列的第i位的第k个氨基酸的频率分布。
- FPt(j,m)是模板序列第j个位置的第m个氨基酸的频率分布图;
- B(k,m)是BLOSUM62评分矩阵,用于将查询的第k个氨基酸与模板的第m个氨基酸对齐。
- 引入常数fshift来平衡新的序列图项,其中c3 = 1.0和fshift = -0.4的值在蛋白质的训练集中确定。
2.1.4 dPPAS and dPPAS2
dPPAS使用以下附加的基于深度的评分功能将查询序列与模板结构对齐
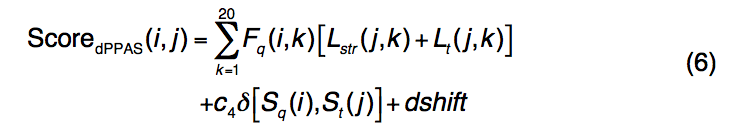
- 在等式(1)中定义了Fq(i,k),Lt(j,k)和δ[Sq(i),St(j)]。
- Lstr(j,k)代表反映模板结构中相邻残基片段的环境和深度的结构轮廓。
为了计算结构轮廓,将每个模板结构分为具有9个残基的小片段,将其用作种子片段,并与由PISCES选择的一组非冗余PDB蛋白中的9个残基片段进行无缝穿线比较,以进行比较。通过RMSD和结构中碎片深度的相似性进行测量。从数据库收集的这些片段用于计算模板的位置特定的频率曲线Lstr(j,k)。 Smith-Waterman局部比对算法用于识别目标序列和模板序列之间的最佳匹配。 dPPAS的参数是根据我们的训练数据集(基于线程模型的TM得分)通过网格搜索来确定的(c4 = 6.5,dshift = -0.96,go = -7.0和ge = -0.54)。 dPPAS2使用与dPPAS相同的评分功能和比对算法,但对序列和深度剖析项的权重更高。
2.1.5 wdPPAS
wdPPAS是dPPAS和wPPAS的扩展,其评分功能由

新的加权配置文件和参数与Eqs(4)和(5)中的相同。 通过Needleman-Wunsch全局动态规划算法搜索比对。
2.1.6 MUSTER
MUSTER(multi-source threader)是一种将序列配置文件与结构预测的不同资源结合在一起的线程算法(threading algorithm )
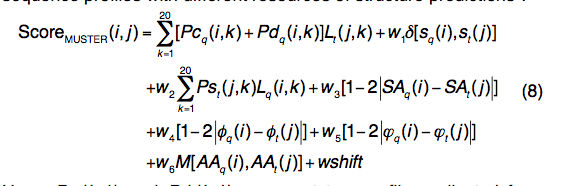
- Pcq(i,k)和Pdq(i,k)分别代表从接近(E值<0.001)和遥远(E值<1.0)同源序列中收集的两个轮廓(profiles)。
- 等式中 (8)的第二和第三项用于二级结构和结构轮廓匹配,类似于等式(6)中用于dPPAS的匹配。
- 第四,第五和第六项用于计算溶剂可及性,查询和模板之间的主链扭转角的匹配。
- 第七项是通用的疏水评分矩阵。
- 设计了一种基于晶格的系统搜索,该搜索旨在最大程度地提高300种非冗余训练蛋白的平均TM-score,从而得到了优化的参数集:w1 = 0.65,w2 = 1.10,w3 = 4.49,w4 = 2.01, w5 = 0.59,w6 = 0.20,wshift = 1.00,go = 6.99和ge = 0.54。
2.1.7 wMUSTER
与MUSTER相比,在wMUSTER评分功能中添加了两个新的加权项:

在这里,PSSpred使用基于PISCES收集的1,192种非冗余PDB蛋白的离散评分矩阵进行了重新训练。 对于每个查询残差,wMUSTER都会考虑由重新训练的PSSpred给出的三个二级结构状态(即H,E和C)和十个置信度得分(即[0、1,…,9])。 STRIDE为模板结构中的每个残基分配了七个二级结构状态之一,即 H:α螺旋,G:3-10 helix,I:PI-helix,E:extended conformation,B:isolated bridge,T: turn , C:coil。 因此,对于不同的PSSpred预测和STRIDE分配对,总共有21个概率得分,每个都有10个置信度级别。 我们计算3×7替换矩阵,每个置信度值分别为
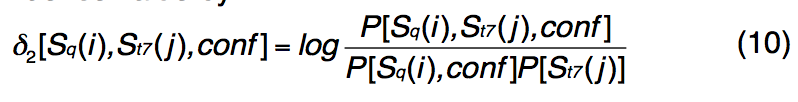
-
其中P [Sq(i),conf]是在置信度得分conf上发生预测的二级结构类型i(i∈[H,E,C])的概率;
-
P [St7(j)]是实际发生次级的概率 结构类型j(j∈[H,E,C,G,B,S,T])。
我们比较了1,192种蛋白质的预测二级结构和实际二级结构,并计算了置信度得分conf和实际二级结构的预测二级结构类型i的联合概率P [Sq(i),St7(j),conf]。 一元结构类型 这样,我们为每个置信度值计算了对数奇数得分函数(3×7替换矩阵)。 等式中的恒定位移。 引入(9)来平衡新项δ2[Sq(i),St7(j),conf]。 与新条款相关的参数 在等式中 (9)是:w7 = 0.5,fshift = -0.5,w8 = 0.5和sshift = -0.8。
2.9 LOMETS的Threading模板组合和目标分类
线程模板(threading templates )和对齐方式由LOMETS组合。 对于程序P中的每个模板,计算Z分数以评估与其他模板相比对齐的重要性:
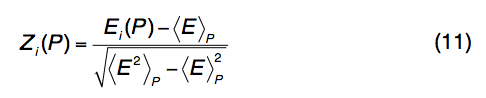
其中Ei(P)是P对齐的分数,而<…> P表示线程程序P在所有模板对齐中的平均值。Z分数Z0(P)分配给每个程序以 根据大型基准测试来区分模板的好坏,即Z0(PP AS)= 7.0,Z0(Env-PP AS)= 8.0,Z0(wPP AS)= 7.0,Z0(dPPAS)= 9.3,Z0(dPPAS2)= 10.5,Z0(wdPPAS)= 9.3,Z0(MUSTER)= 5.8和Z0(wMUSTER)= 5.8。
如果有超过八个模板的Zi(P)> Z0(P),即每个线程程序好的模板的平均数量> 1,则将目标定义为通常对应于同源蛋白质的Easy目标。 如果没有Zi(P)> Z0(P)的模板,则将其定义为通常对应于非同源蛋白的硬靶标。 其他是中等目标。
生成一个“ init.dat”文件,该文件从每个线程程序中收集前20个模板。 “ init.dat”中的模板按良好对齐的顺序列出,然后按不良对齐的顺序列出,从高置信度程序到低置信度程序枚举,其中,线程程序的置信度由对齐的平均TM得分排名 在大型基准测试中。 I-TASSER将使用此文件生成空间约束并构建装配模拟开始的初始构象。
2.10 I-TASSER中蛋白质结构的构象表示
I-TASSER中的每个残基均由其Cα原子和侧链质心(center)表示。 根据LOMETS的查询到模板的比对,查询序列被分为线程的对齐区域和未对齐区域。 从长度大于5的模板结构的连续件中切出螺纹对齐区域中的构象。 这部分序列的局部结构在仿真过程中保持冻结状态,并在非网格系统上移动。 它旨在增强模板对齐方式可提供的高分辨率结构的保真度(图A)。
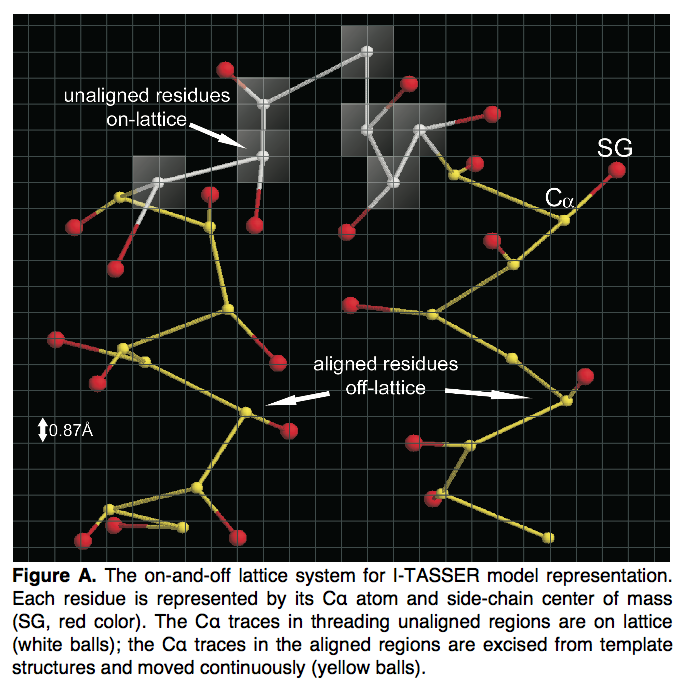
threading未对齐区域的构象模拟是在基于网格的系统上进行的,该系统的网格比例为0.81 A(图A中的白色区域);这旨在减少构象搜索熵( con- formational search entropy)。为了进一步加快构象搜索速度,我们定义了312个Cα-Cα晶格键矢量,长度在3.25至4.35 Å之间,其中所有允许的2键组合均通过排除任何带有低于65°或高于165°的键角,这在PDB结构中从未见过。该过滤器可以大大减少晶格上的搜索空间。晶格上键矢量的平均长度为3.81 A ,与PDB结构中看到的标准Cα-Cα键长一致。但是为了增加晶格运动的构象灵活性,单个键的长度可能在3.25至4.25 A 之间变化。
I-TASSER模型的拓扑由各个线程片段的相对方向决定,其中未使用的格子区域是从头开始构建的,这些区域用作线程片段的链接。
2.11 I-TASSER movements和蒙特卡洛搜索引擎
I-TASSER中的结构重组是通过副本交换蒙特卡洛(replica exchange Monte Carlo, REMC)模拟进行的[17,18]。I-TASSER模拟中有两种类型的构象运动(图B)。

第一种晶格运动包括六组局部运动:(A1)2键矢量行走( 2-bond vector walk;); (A2)3键矢量行走; (A3)4-键向量步; (A4)5键向量行走; (A5)6键向量步态;(A6)N或C端随机步态(图B)。 为了加快仿真速度,预先计算了跨越移动窗口的任何给定距离矢量的所有2键和3键构象,因此可以通过枚举运动查找表来快速进行A1和A2运动 无需计算特定的残基坐标。 还可以通过A1和A2运动的组合来快速地进行A3-A5运动,即通过沿移动窗口顺序枚举相邻的2-或3-键矢量。
第二种非晶格运动由随机选择的连续片段的四组全局运动组成:(B1)片段的平移; (B2)使片段围绕随机选择的残基旋转; (B3)片段旋转和翻译的结合; (B4)通过独立旋转片段的N和C端产生小的变形。 片段每次脱离晶格运动后,将重新生成两端的相邻晶格矢量以保持链连接(图B)。
遵循标准REMC协议17,存在并行执行的仿真的Nrep副本,第i个副本的温度为
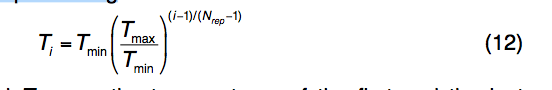
其中Tmin和Tmax分别是第一个和最后一个副本的温度。 Nrep范围从40到80,Tmin从1.6到1.98 kb-1,Tmax从66到106 kB,这取决于蛋白质的大小,其中较大的蛋白质具有更多的复制品和更高的温度。 对于不同大小的蛋白质,这些参数设置可导致最低温度复制品的接受率为〜3%,最高温度复制品的接受率为〜65%。
在每200 * L构象运动之后,将按照标准Metropolis准则尝试每对相邻副本之间的全局交换运动,概率为〜exp(ΔβΔE),其中等式中的温度分布为,在上述温度和Nrep参数设置下,方程(12)中的温度分布导致所有相邻副本之间的交换运动的接受率大约为40%。
2.12 I-TASSER力场构造
I-TASSER模拟中未对准区域和对准区域的模拟均由相同的基于知识的统一力场控制,该力场建立在简化模型上,每个残基均由Cα原子和碳原子的侧链中心指定 质量 I-TASSER力场由以下三个主要部分组成。
(1)一般统计潜力 Generic statistical potentials
通用的统计潜能来自PDB库中观察到的结构规律:
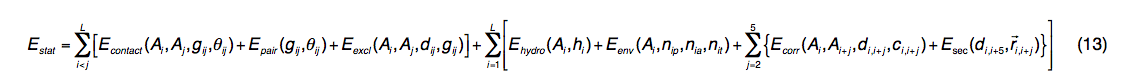
其中L是查询长度; dij和gij分别是第i个和第j个残基之间的Cα和侧链中心距离,其中Ai和Aj是残基的氨基酸同一性。 θij表示接触的两个残基(i和j)的侧链向量的方向,其值分为三组(平行,反平行和垂直)。
Econtact(Ai,Aj,gij,θij)是从6,500种非冗余高分辨率PDB结构派生而来的特定于方向的通用接触电势。 Epair(gij,θij)的导出方法与Econtact相同,但是contact的计数是通过查询的残基对与±5个相邻残基窗口上的PDB结构之间的BLOSUM变异分数之和加权的。。 这种潜力是特定于查询序列的,但是查询和PDB结构之间不需要对齐,因为我们计算了与查询具有相同氨基酸同一性(Ai,Aj)的PDB结构中的所有接触对。 Eexcl(Ai,Aj,dij,gij)表示基于1 / dij(或1 / gij)的依赖关系,Cα和侧链中心之间的软核排除体积电势。
Ehydro(Ai,hi)是基于通用Kyte-Doolittle亲水性基质的得分和通过神经网络训练预测的取决于序列的溶剂可及性的疏水相互作用,其中hi是溶剂中第i个残基的深度。
Eenv(Ai,nip,nia,nit)是特定于取向的接触曲线,其中nip,nia和nit是与第i个残基接触的残基数,侧链向量是平行,反平行或垂直分别对应第i个残基的残基。
Ecorr(Ai,Aj,di,i + j,ci,i + j)是第i个和第(i + j)个残基之间的短距离Cα距离相关性,其中ci,i + j表示局部的手性结构体。
Eesc(d,ri,j_j)是由di,i + 5指定的二级结构倾向,即局部结构将被调节为α-螺旋(或β-链),当di,i + 5低于7.5 A(或高于11 A)时,二级结构规则性由Cα距离和基于标准α-的参数的相邻Cα键矢量(r i,j+1)的相对方向表示螺旋结构和β链结构。
等式(13)中的许多术语已在Refs.7,20,21中进行了描述 . 这些条款中使用的参数可从 http://zhanglab.ccmb.med.umich.edu/potential/ 下载。
(2)氢键网络。
I-TASSER中的氢键(H键)由遵循PSSpred二级结构预测的骨架几何结构指定。 通过接触顺序(CO,沿序列的残基距离)以及供体和受体残基的相对方向和距离来评估模拟中氢键的出现(图C)。 例如,仅当CO = 3且供体和受体残基(bb和cc)的平分线与法向向量的乘积大于特定阈值且Cα距离(r)以上时,α-螺旋中的氢键才会出现 低于临界值; 考虑到满足相同的H键几何截断要求,对于反向平行或平行的β-折叠,氢键残基的CO分别要求> 4或> 20。
氢键能由下式计算
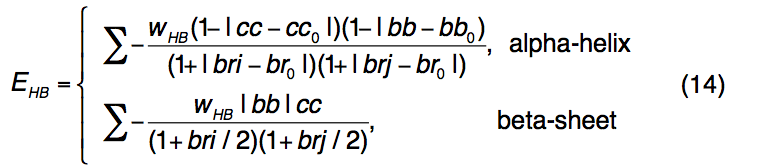
用来惩罚候选H键与标准H键几何形状的结构偏差。 如果供体和受体残基均被预测为α-螺旋或β-立场,则 wHB = 1; 否则,wHB = 0.5。 标准氢键的截断参数(cc0,bb0,br0)是根据500个高分辨率PDB结构的平均值计算得出的,其中二级结构元素由STRIDE分配。

(3)基于线程模板的约束
基于线程的空间约束包含Cα距离图和侧链接触,它们通过从LOMETS获取的顶级Ntemp线程模板的共识从’init.dat’文件中收集,其中Ntemp = 20表示Easy目标,“中等”目标为30,“硬”目标为50,遵循LOMETS目标(target)定义后。 在第二轮迭代模拟中,除了线程约束之外,外部约束还包括I-TASSER簇质心的约束和使用该簇的结构对齐程序TM-align检测到的PDB模板的约束,聚类结构作为探针。
通过以下方式在I-TASSER中实施四种约束

其中dij和gij是第i个残基和第j个残基之间的Cα和质点侧链中心; dp是两个Cα原子之间的预测距离ij; g0Ai Aj是侧链接触的氨基酸比距离截止值; θ(x)是阶跃函数,当x≥0时等于1,当x <0时等于0; 当约束(confij)的置信度得分高于阈值(confcut)时,w(confij)= 1 + | confij-confcut | 4,否则,w(confij)= 1- | confij-confcut |2。
I-TASSER能量项通过线性回归进行组合,其中通过最大化诱饵的总能量和TM得分与原始状态之间的相关性,在一组100×60,000诱饵上优化不同项的加权参数。
2.13 结构模型选择和原子级细化 Structural model selection and atomic-level refinement
由SPICKER群集程序选择I-TASSER结构装配模拟中最频繁出现的构象。 这些构象对应于蒙特卡洛模拟中最低自由能态的模型,因为每个构象簇nc的诱饵数与分配函数Zc成正比,即 $ n_{c} ~ Z_{c} = ∫ e ^{-βE}dE $ 。 因此,归一化簇大小的对数与模拟的自由能有关,即
$ F = -k_{B} T logZ〜log(n_{c} / n_{tot}) $
,其中ntot是提交聚类的诱饵的总数。
每个SPICKER群集中的质心模型是通过平均所有群集构象的坐标生成的。 由于质心模型通常包含空间碰撞,因此I-TASSER进行了第二轮装配仿真,以消除局部碰撞并进一步完善全局拓扑。 在第二轮重组模拟中,通过使用TM-align搜索PDB库中与第一轮模拟中的簇质心相似的结构,从检测到的结构模板中添加空间约束。
最终的原子模型由ModRefiner从第二轮仿真轨迹中选择的低能构象开始构建。 在ModRefiner中,首先从Cα迹线构建主干结构。 然后从旋转异构体库中构建侧链原子,并基于基于物理学和知识的复合力场,通过最小化能量来完善全原子构象。
2.14 I-TASSER结构预测的全局质量估计
并非所有的结构预测都是准确的。 因此,建模准确性的估算对于决定用户在自己的研究中应如何利用模型至关重要。 I-TASSER结构模型的准确性是通过计算结构装配模拟的置信度得分(或C分数)来估算的:
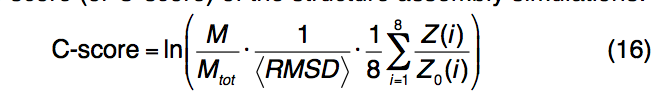
其中M是SPICKER群集中结构的多样性; Mtot是集群中使用的I-TASSER结构诱饵的总数; 〈RMSD〉是诱饵对簇质心的平均RMSD。这些术语对应于结构装配模拟的收敛程度。 Z(i)和Z0(i)是第i个LOMETS线程程序的模板的最高Z分数,以及用于区分好模板和坏模板的相应Z分数临界值,如第8节中所定义。标准化Z分数测量threading对齐的重要性,并与LOMETS模板的质量相对应。
大规模基准测试表明,C得分与I-TASSER模型的准确性之间存在很强的相关性。 C分数与实际TM分数之间的相关系数为0.91,C分数与RMSD之间的相关系数为0.75。这些数据允许对与原始状态有关的第一个预测模型的TM得分和RMSD进行定量估计:

其中C是C-score,L是目标序列的长度。 在针对500种非冗余蛋白的大规模基准测试中,使用Eq(17)进行质量评估的平均误差对于TM评分和RMSD分别为0.008和2.0Å。
2.15 I-TASSER结构预测的残基水平局部质量估计
根据建模仿真的变化和同源比对的不确定性,开发了一种算法(ResQ)来估计I-TASSER预测的残基级局部质量。 为了训练该方法,我们首先对来自PDB的1,270种非冗余单域蛋白进行了基于I-TASSER的结构预测,这些蛋白随机分为两组训练蛋白和测试蛋白。 与原始结构相比,支持向量回归(SVR)用于训练I-TASSER模型的残基特定距离误差,涉及以下五个特征。
(1)装配模拟的结构变化 Structural variation of assembly simulations.
REMC模拟中第j个残基的结构变异由平均值和标准偏差定义:
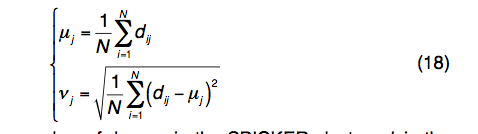
其中N是SPICKER群集中的诱饵数量; dij是在TM得分叠加之后第i个诱饵结构与质心结构模型之间的第j个残基的距离。一般而言,具有较高变异性的残基相对于天然残基具有较大的误差,反之亦然。
(2)最终模型与基于序列的特征预测之间的一致性。目标序列的二级结构(SS)和溶剂可及性(SA)由PSSpred和SOLVE(Zhang等人,未公开)程序预测,将其与STRIDE程序分配的3D结构模型的实际SS和SA进行比较。模型和预测之间SS和SA不一致的残基通常具有较大的误差。
(3)Threading对齐coverage。残基的对齐覆盖率定义为具有查询残基对齐的Threading模板的数量除以模板总数除以LOMETS。具有较高线程覆盖率的残渣表明在模拟过程中对它们的约束更多,并且大概具有较高的建模精度。
(4)LOMETS线程处理模板的结构变化。考虑到LOMETS15将Nj模板与查询序列上的第j个残基对齐,则LOMETS线程模板的结构变化定义为:
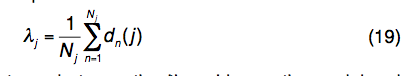
其中dn(j)是模型上的第j个残差与 第n个模板上与查询中第j个残基对齐的残基。距离是在将模板结构通过TM得分旋转矩阵叠加在查询模型上后计算得出的。如果Nj为零,则λj的值设置为10Å。
(5)TM-align结构比对中模板的结构变异。 TM-align将查询结构模型针对非冗余PDB库进行处理,以查找共享相似拓扑的模板。 TM-align模板的结构变化的定义与方程式(19)相同。 但TM对齐模板定义了Nj和dn(j)。 从I-TASSER模型和模拟诱饵开始,ResQ为每个残基生成距离误差估计。对I-TASSER C得分> -1.5的506种非冗余蛋白进行的基准测试表明,与X射线晶体学数据相比,可以估计残基水平的准确度,平均误差<1.5Å。
2.16 B因子估算 B-factor estimation
B因子与局部原子和残基的固有热迁移率有关,这对于蛋白质在生理环境中折叠和发挥功能至关重要。 B因子预测由SVR在基于模板的分配和PSI-BLAST配置文件上进行训练。
(1)基于模板的分配。 I-TASSER根据通过LOMETS和TM-align从原始PDB条目中提取的顶级同源和类似模板的实验B因子值,为每个查询残基分配B因子。

其中nj是残基对齐的模板数 在查询残基j上,bt(i,j)是第i个模板的残基的标准化B因子值,该模板通过LOMETS和TM-align在j上对齐。
(2)Sequence profiles。 PSI-BLAST1通过NCBI非冗余序列数据库搜索靶序列,以检索同源序列,这些序列以位置特异性得分矩阵(PSSM)的形式表示。对于每个残基,在将其元素x在(0,1)范围内转换为[[1 + exp(-x)]]后,使用大小为9个残基的滑动窗口从PSSM中提取轮廓特征。源自PSI-BLAST序列图谱的二级结构和溶剂可及性,也用作B因子预测的特征。使用序列图谱的假设是,保守性更高的残基通常在结构上更稳定,因此具有较低的B因子,反之亦然。
对635种非冗余蛋白进行的基准测试表明,ResQ估计的B因子与X射线晶体学数据的皮尔森相关系数为0.60。
2.17 BioLiP:用于基于结构的蛋白质功能注释的复合数据库
PDB库是蛋白质结构和功能分析研究的主要来源。但是PDB中的许多蛋白质都包含多余的条目,残基排序错误和功能注释错误。此外,使用人工分子作为添加剂可解析许多蛋白质结构,以利于结构确定。这些错误和人为添加物阻碍了该文库成为进行精确的基于结构的功能分析的可靠资源,这需要开发经过仔细验证的生物学功能的清洁蛋白质文库。我们提出了由多个计算机过滤和人工文献验证组成的分级程序,以评估PDB中共结构配体的生物学相关性。一个全面的功能数据库BioLiP是从PubMed中已知的结构/功能数据库和文献中构建的,每个条目都包含配体结合位点,结合亲和力和催化位点的注释。
BioLiP每周更新一次,并可以从 http://zhanglab.ccmb.med.umich.edu/BioLiP 免费向社区提供。 。当前的BioLiP版本(2014年10月31日)包含来自64,287个独特蛋白的298,556个配体结合条目,其中DNA / RNA蛋白为37,223个,肽蛋白为13,498个,金属离子蛋白为84,248个,小蛋白为163,587个分子-蛋白质相互作用。从公共数据库和人工文献调查中收集的实验结合亲和力数据总计23,492个。
最近,BioLiP进行了扩展,以集成UniProtKB,酶委员会(EC)和基因本体论(GO)中基于序列的功能注释,其中序列数据库中的EC和GO信息通过严格的序列映射到BioLiP结构条目 通过手动验证进行对齐。 它涉及75,462条蛋白质链,其中513个唯一的前3位数字和2,413个唯一的4位数字酶委员会(EC)号,以及37,178条具有已知的催化残基的链,其衍生自Catalytic Site Atlas(CSA)。 它还包含119,004条链/结构域,与8,315个独特基因本体论(GO)术语相关。 这些数据为I-TASSER Suite中基于结构的功能注释提供了基础资源。
2.18 COFACTOR用于配体结合位点(LBS)预测 COFACTOR for ligand-binding site (LBS) predictions
COFACTOR通过映射从BioLiP文库中配体-蛋白质复合物的已知结构获得的LBS来预测LBS,该结构与查询结构模型具有相似的结构。 通过将整体结构对齐和局部几何细化相结合,可以检测出结构上的相似性。
对于全局结构对齐,TM-align用于标识与查询折叠相似的模板,即TM-score > 0.5。对于局部几何对齐,使用迭代Needleman-Wunsch动态编程,将模板结合位点区域与查询片段对齐,这些查询片段是基于MSA保守性从查询结构中提取的。 查询蛋白和模板蛋白之间的局部结构和序列相似性(Lsim,local structural and sequence similarity)通过
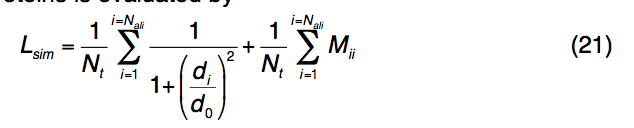
其中Nt代表以已知结合位点为中心的球体中残基的总数,Nali是球体中查询模板对齐的残基对的数量,di是第i个对齐的残基对之间的Cα距离,d0是选择的距离截止为3.0Å,Mii是第i对残基之间的标准化BLOSUM62替代得分。对于模板上的每个binding pocket,将对所有保守的查询模体执行此过程,并记录Lsim最高的那个。模板配体根据局部结构排列的旋转和平移矩阵叠加到查询结构上。最后,所有基于模板的配体构象都基于空间接近度以8Å的距离进行聚类。如果结合口袋与多个配体结合(例如,ATP结合口袋也可以结合MG,PO4和ADP),则基于平均化学键合,基于相同的化学相似性(Tan-imoto系数截止= 0.7)将同一口袋中的配体进一步聚类聚类过程对预测的结合位点进行排序。
最终从每个簇中选择具有最高配体结合置信度得分(CSc,ligand-binding confidence score)的蛋白质-配体复合物,作为查询蛋白质的功能位点预测,即
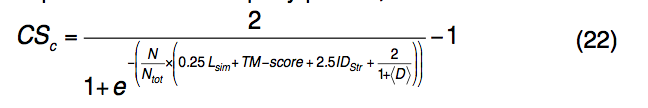
其中N是簇中模板配体的数量,Ntot是使用模板预测的配体的总数。 IDstr是查询和模板在结构上对齐的区域之间的序列同一性。 <D>是同一簇中预测配体与所有其他预测配体的平均距离。
2.19 TM-SITE用于配体结合位点预测 TM-SITE for ligand-binding site predictions
TM-SITE是从COFACTOR扩展的另一种基于结构的功能注释算法,用于通过结构相似性定位已知蛋白质的LBS。 与组合全局和局部结构比对的COFACTOR不同,TM-SITE通过考虑查询和模板蛋白上从第一个结合残基到最后一个结合残基(称为SSFL)的子序列的结构相似性来检测功能同源性。
为了识别功能同源性,TM-SITE使用TM-align将查询结构与BioLiP中预先计算的模板SSFL进行比对。 每对查询和模板SSFL结构之间的匹配由一个综合评分函数评估,该评分函数计算全局和局部以及结构和序列的相似性:
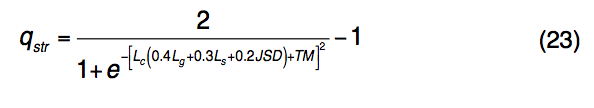
其中Lc是通过TM-align与查询结构对齐的模板结合残基的分数。
 解释了查询蛋白和模板蛋白的结合口袋之间的局部结构相似性,其中n是与模板的结合口袋相关的对齐残基对的数目,di是第i个残基对的距离。
解释了查询蛋白和模板蛋白的结合口袋之间的局部结构相似性,其中n是与模板的结合口袋相关的对齐残基对的数目,di是第i个残基对的距离。
$ L_{s} = \frac{1}{n}\sum_{i=1}^{n} B(R_{i}^{q},R_{i}^{t})$ 测量对齐的结合残基对之间的进化关系, $ B(R_{i}^{q},R_{i}^{t})$ 是第i个对齐残基对的标准化BLOSUM62得分。JSD是一种进化保守性指数,定义为对预测结合残基的平均詹森-香农散度得分(Jensen–Shannon divergence score ),该得分是根据多个序列比对计算得出的。
$ TM= 2TM_{t} * TM_{q}/ (TM_{t} + TM_{q})$ 是对齐查询和模板SSFL时TM-align返回的两个TM得分的谐波平均值(harmonic average ),其中TMt和TMq是分别由查询和模板长度归一化的TM分数
为了从多个SSFL模板中选择LBS残基,根据TM-align的相应比对,将与假定模板库中与蛋白质结合的所有配体投射到查询结构。 这些配体基于它们的几何中心之间的空间距离进行聚类,在这种情况下,平均连锁聚类算法(average linkage clustering algorithm)的距离截止值为4Å。 对于每个簇,基于最大投票从簇中的所有配体推导一组通常对应于一个结合口袋的共有结合残基。 票数> 25%的残基被认为是结合口袋中最终预测的LBS残基。 与特定配体结合簇相关的预测结合残基的置信度得分定义为
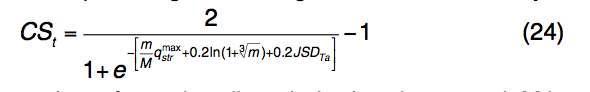
其中m是簇中模板配体的数量,M是所选模板的总数。 $ q_{str}^{max}$是根据等式(23)计算的来自群集模板的qstr得分的最大值。 JSDTa是配体簇中预测的LBS残基的平均JSD得分。 在最终的TM-SITE预测中,将根据CSt分数选择并对其进行排序,并根据每个结合口袋中的投票数对结合残基进行排序。
2.20 S-SITE用于配体结合位点预测 S-SITE for ligand-binding site predictions
S-SITE是I-TASSER包装中的第三个基于模板的方法,它使用结合位点特异性,序列图谱-图谱比较来检测蛋白质功能模板和配体结合位点。为了获得查询蛋白的图谱,使用PSI-BLAST将查询序列穿入NCBI NR序列数据库,以构建多个序列比对,类似于PSSpred和LOMETS中使用的过程。然后从多个序列比对中计算出位置特定的频率矩阵(PSFM)。模板图谱由位置特异性评分矩阵(PSSM)表示,并由PSI-BLAST搜索在BioLiP库中的所有蛋白质上预先构建。为了从BioLiP中检测同源模板,使用Needleman-Wunsch动态编程算法将查询配置文件PSFM与库中的模板配置文件PSSM进行比较,得分将查询中的第i个残基与定义的模板中的第j个残基对齐如
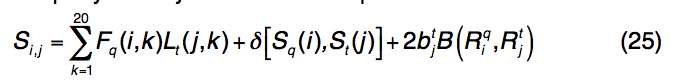
公式(25)中的前两个术语描述了轮廓-轮廓对齐和二级结构匹配,类似于公式(1) 。 第三个术语描述了结合口袋残基的进化保守性,如果第j个残基在模板的结合位点,则btj = 1,否则,bt = 1。 $ B(R_{i}^{q},R_{j}^{t})$ 是查询中的残基 $R_{i}^{q}$和模板中的 $R_{j}^{q}$残基的标准化BLOSUM62相似性评分,其中值为[0,1]。 S-SITE中模板匹配的质量估算为
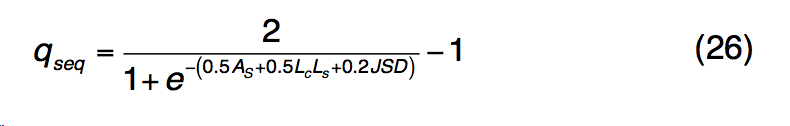
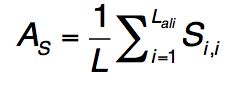 是通过查询序列长度L归一化的轮廓比对得分。Lc,Ls和JSD与等式(23)中定义的相似,但具有从配体结合特异性产生的比对 profile-profile 比较。 qseq得分高于0.5的BioLiP中的所有蛋白质均被选为推定模板。 如果假定模板的数量少于10个,则将返回qseq得分最高的前10个模板,以进行下一步LBS选择分析。 在序列图谱-谱图比对之后,与模板上的结合残基对齐的查询残基在S-SITE预测中被指定为推定的结合残基。 由于不同模板的结合位点将与不同的查询残基匹配,因此采用共识投票方案选择最一致的结合残基。 S-SITE将获得> 25%投票的残基视为最终结合残基。
是通过查询序列长度L归一化的轮廓比对得分。Lc,Ls和JSD与等式(23)中定义的相似,但具有从配体结合特异性产生的比对 profile-profile 比较。 qseq得分高于0.5的BioLiP中的所有蛋白质均被选为推定模板。 如果假定模板的数量少于10个,则将返回qseq得分最高的前10个模板,以进行下一步LBS选择分析。 在序列图谱-谱图比对之后,与模板上的结合残基对齐的查询残基在S-SITE预测中被指定为推定的结合残基。 由于不同模板的结合位点将与不同的查询残基匹配,因此采用共识投票方案选择最一致的结合残基。 S-SITE将获得> 25%投票的残基视为最终结合残基。
为S-SITE中的结合残基定义了置信度得分CSs:
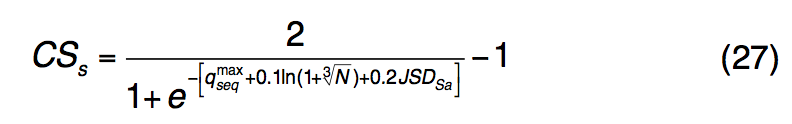
$q_{seq}^{max}$是所有假定模板中qseq的最大值,N是所选模板的数量,JSDSa是所有预测的LBS残基的平均Jensen-Shannon divergence评分。 在此,置信度得分CSs的格式与TM-SITE(CSt)相似,但是在S-SITE中不进行聚类,因为从序列概况比较中检测到的模板在大多数情况下是会聚的。
2.21 COACH用于共有配体结合位点预测 COACH for consensus ligand-binding site prediction
COACH是一种用于LBS预测的共识方法,它使用线性支持向量机(SVM)结合了COFACTOR,TM-SITE和S-SITE的算法的预测结果(图D)。为了生成LBS预测,查询序列与I-TASSER结构模型一起作为输入提供,并馈送到各个程序中。残基成为LBS残基的可能性是通过各种方法计算的,这些方法用作残基SVM的输入特征向量。 COFACTOR的概率取自默认置信度得分(公式22中的CSc)。对于TM-SITE和S-SITE,将概率计算为配体簇或模板(即等式24和27中的CSt和CSs)的置信度分数乘以残基的投票比率。最后,将所有特征向量馈入SVM进行共识预测,并使用PISCES收集的分类器对400种非冗余训练蛋白进行训练。
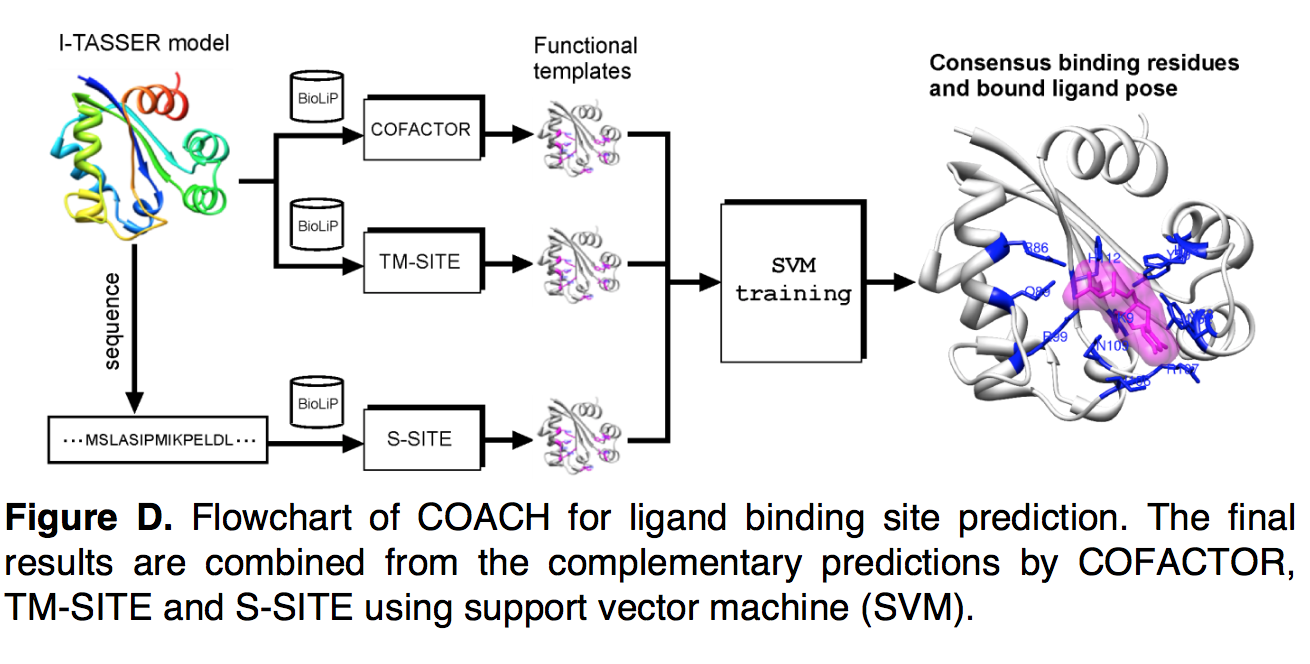
使用SVM-light中的线性核(linear kernel),并基于详尽的网格搜索选择了成本参数(cost parameter) C的最佳值。 为了避免过度训练,在COACH中采用了10倍交叉验证程序,即将训练集随机分为10个大小相等的子集,其中9个子集用于训练SVM,其余子集用作验证 计算LBS预测的平均Matthews相关系数(MCC)。 对于网格空间中的每个参数C,将这种随机样本划分重复10次,并计算总MCC作为10个MCC的平均值。 最终选择了具有最高总体MCC的参数C进行COACH中的SVM训练。
2.22 基于结构的酶功能预测
酶是在生理过程中催化化学反应的蛋白质。 根据它们催化的反应,使用称为酶委员会(EC, Enzyme Commission )编号的数字分类方案将酶分类为等级族。 为了从氨基酸序列预测EC数,我们的方法涉及将预测的蛋白质结构与全局和局部3D酶模板进行结构匹配。 EC预测的置信度得分EC得分定义为
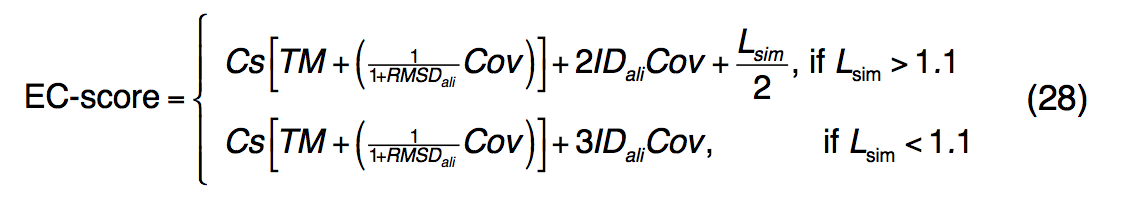
其中Cs是等式(16)中定义的I-TASSER预测的C-score。 在将目标I-TASSER模型与BioLiP中的模板结构进行比较时,TM,RMSDali,Cov和IDali是TM序列返回的TM得分,RMSD,比对范围和序列同一性。 Lsim是查询和模板绑定袋之间的本地活动站点匹配分数,如公式(21)中所述。
为了生成用于查询的初始结合袋,通过查询序列扫描模板的已知催化/活性残基。 氨基酸类型与模板上催化/活性残基相似的残基在查询中标记为潜在的活性位点。 模板的结合型口袋球定义为以模板的活动位点的几何中心为中心并包含<30个残基(或半径<20Å)的球。 结合口袋的优化比对通过类似于COFACTOR和TM-align中使用的启发式迭代程序进行。
2.23 基于结构的基因本体预测
基因本体论(GO)是一种广泛使用的词汇,用于描述基因功能的三种不同分类法或“方面”(aspects):分子功能,生物过程和细胞成分。每个GO方面都表示为一个结构化的有向无环图,其中图中的节点代表GO项并描述基因产物功能的组成部分,而节点之间的边等于关系(' -a’或’part-of')。 GO术语以功能层次结构的形式保存,其中更通用的功能位于顶部,而更具体的功能位于图表的下方。
为了识别GO术语和查询蛋白的功能成分,我们使用了一种类似于LBS和EC预测的基于结构的方法。由于可以通过TM比对结构比较来识别多个模板,因此将GO预测的可信度GO分数定义为所有具有相同模板的序列和结构相似性评分(SaS)的归一化总和GO条款:
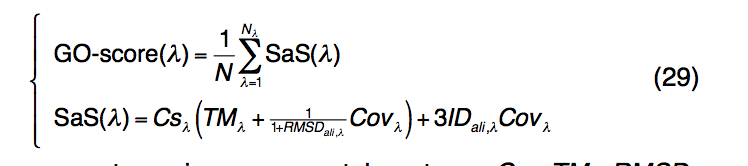
其中,λ代表给定的基因本体项。 Csλ,TMλ,RMSDλ,Covλ,IDali,λ与等式(28)中定义的术语相似,但带有特定的GO标记λ。 Nλ是与GO项λ相关的模板的数量,N是为生成共识GO预测而选择的模板的总数。当有多个同源模板可用时,我们仅考虑SaS得分> 1的模板。对于那些少于10个SaS得分模板大于1的查询蛋白,无论SaS得分如何,都选择前10个模板来生成共有预测。一旦识别出GO得分高于GO得分临界值的GO项,就消除了有向无环图(DAG)中所有其祖先项,以避免冗余。最近对700种非冗余蛋白进行的大规模基准测试表明,基于I-TASSER模型的EC和GO预测的准确性明显高于基于序列的(PSI-BLAST)和基于线程的( MUSTER和HHpred)方法。
2.24 I-TASSER套件的安装和实施
(1)安装I-TASSER
I-TASSER Suite是在Linux操作系统上开发的,其核心程序是用FORTRAN,C / C ++和Java语言编写的。 可以从 http://zhanglab.ccmb.med.umich.edu/I-TASSER/download/ 获得该软件包。 每周更新的结构和功能库可从 http://zhanglab.ccmb.med.umich.edu/library 获得,软件包中提供了Perl脚本“ download_lib.pl”,用于自动库下载。 该软件包包括一个程序(‘update_IT_lib.tar.bz2’),用户可以使用该程序自行根据任何蛋白质结构创建模板文件。 图E提供了一个屏幕快照,该屏幕快照说明了安装后I-TASSER软件包的结构。

(2)I-TASSER实施 I-TASSER implementation
为了解释I-TASSER Suite的实现,我们假设包,模板库,java可执行程序和输入文件分别位于$ pkgdir,$ libdir,$ java / bin / java和$ datadir。 这些目录可以安装在用户计算机上的任何文件夹中。 运行I-TASSER的主要脚本位于“ $pkgdir/ITASSERmod/runI-T ASSER.pl”。 不带参数运行“ $pkgdir/I-TASSERmod/runI-TASSER.pl” 可以打印出各种选项的简要帮助信息。 运行I-TASSER的一个示例命令是
$pkgdir/runI-TASSER.pl -pkgdir $pkgdir -libdir $libdir – runstyle serial -LBS true -EC true -GO true -seqname example -datadir $datadir –outdir $outdir -java_home $java - light true -hours 6
在此示例中,I-TASSER以由选项“ –runstyle serial”指定的串行模式运行,查询蛋白名为“ example”。 将针对3D结构预测和生物学功能注释(包括配体结合位点(LBS),酶委员会编号(EC)和基因本体论术语(GO))进行建模。 每个I-TASSER模拟作业的最大运行时间设置为6小时,这由选项“ -light true”和“ -hours 6”指定。 用户还可以在并行模式下运行I-TASSER,该模式由选项“ –runstyle parallel”指定,在该模式下,线程和I-TASSER模拟作业将由作业调度程序使用“ qsub”提交到计算机节点集群中。 命令(假设已安装PBS Job Scheduler系统)。 并行模式可以显着加速I-TASSER建模。
(3)输入数据准备
运行I-TASSER Suite所需的唯一输入信息是目标蛋白质的氨基酸序列,该序列必须保存在 $datadir文件夹中名为“ seq.fasta”的文件中。 图F显示了输入示例。 除了序列信息外,用户还可以使用以下四个选项之一指定其他约束和模板以指导I-TASSER建模:-restraint1,-restraint2,-restraint3,-restraint4。 用户必须首先在$ datadir中创建所有约束文件,然后根据选项指定文件名(请参阅 http://zhanglab.ccmb.med.umich.edu/I-TASSER/download/README.txt )。
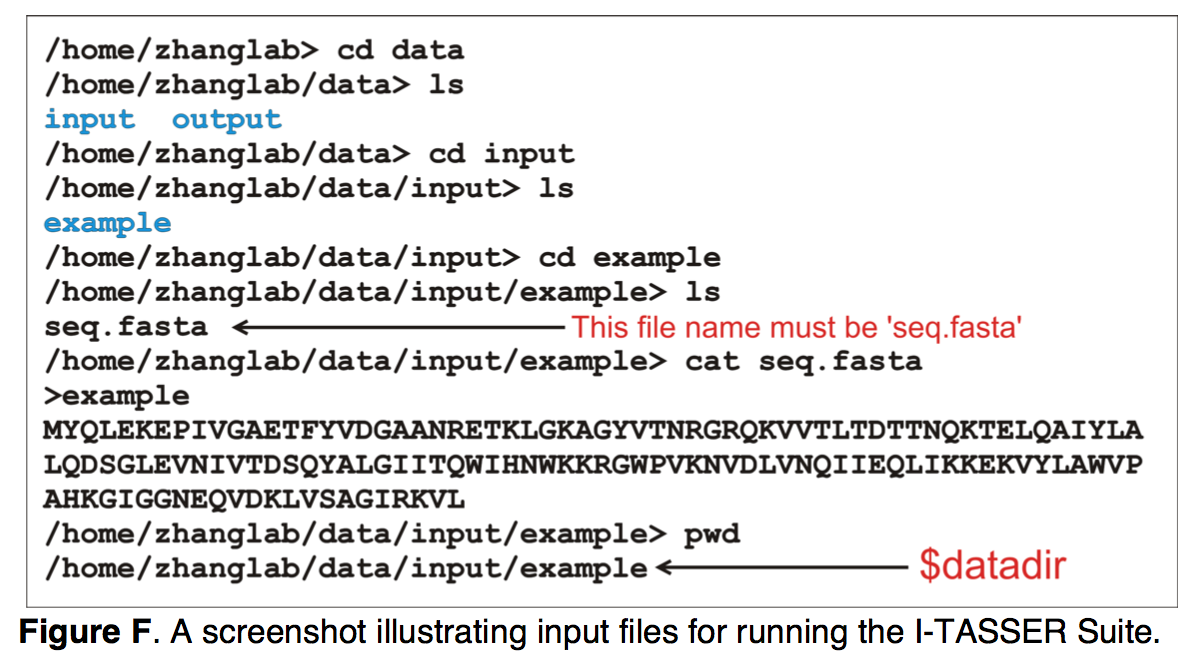
(4)解释结构预测结果 Interpreting structure prediction results
I-TASSER结构建模的输出结果位于$ outdir文件夹中,该文件夹由选项“ –outdir”指定。 如果未指定输出目录,则默认文件夹$ outdir = $ datadir。 结构建模输出包括线程模板,仿真轨迹文件,最多五个结构模型和置信度得分估计。
图G显示了基于I-TASSER的结构建模输出结果的屏幕截图,其中补充表1总结了I-TASSER结构预测的输出文件的注释。

(5)解释功能预测结果
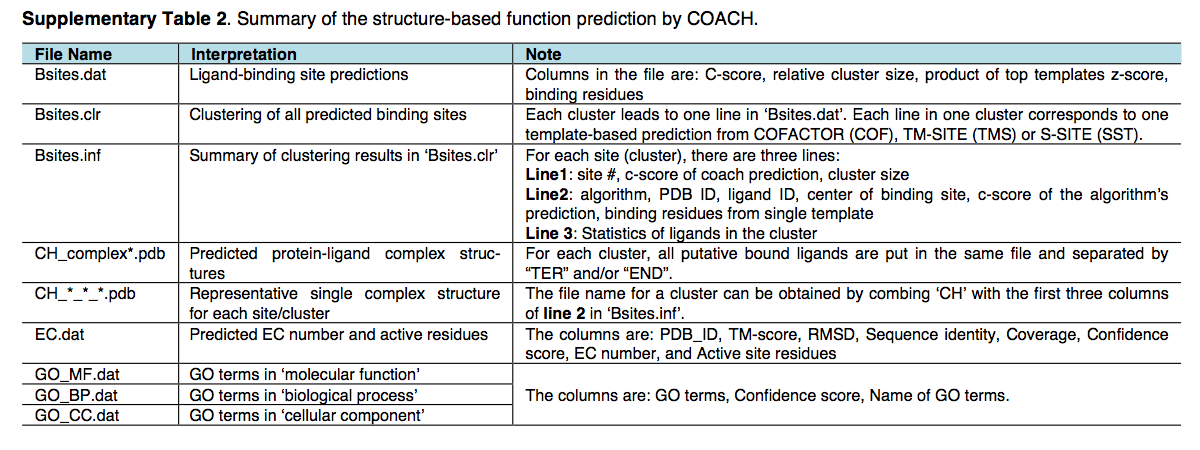
COACH基于结构的功能预测的结果位于 $out-dir/model1/coach,其中文件项在图H和补充表2中进行了解释。除了一致的COACH结果之外,功能注释 由TM-SITE,S-SITE和COFACTOR的各个预测变量分别列出在 $outdir/model1/tmsite,$out-dir/ssite 和 $outdir/model1/cofactor/ 中。

参考资料
- NATURE METHODS | VOL.12 NO.1 | JANUARY 2015 | 7. The I-TASSER Suite: protein structure and function prediction
