【4.5.1】抗体编号系统(Kabat/Chothia/IMGT/Gelfand/Aho/Martin )以及CDR定义
这篇来自文献《Understanding the Significance and Implications of Antibody Numbering and Antigen-Binding Surface/Residue Definition》,实在是太棒了,忍不住一字一句的来理解。
单克隆抗体在人类和动物健康中发挥着越来越重要的作用。蛋白质和化学工程的不同策略,包括非人抗体的人源化技术,成功应用于优化抗体的临床表现。尽管出现了允许开发完全人抗体如转基因Xeno小鼠的技术,但抗体人源化仍然是治疗性抗体的标准方法。抗体人源化的重要先决条件需要标准化的编号方法来精确定义互补的决定区(complementary determining regions,CDR),框架( frameworks ),影响抗体 - 抗原相互作用的结合亲和力和特异性的轻链和重链的残基。最近生成的深度测序数据和来自人类和非人类来源的抗体的解析三维结构的数量不断增加,导致出现了大量数据库。但是,这些不同的数据库使用不同的编号约定和CDR定义。此外,可变链长的大幅波动,特别是在重链CDR3(CDRH3)中,使抗体序列的比较和分析以及抗原结合残基的鉴定复杂化。本综述比较和讨论了迄今为止建立的不同编号方案和“CDR”定义。此外,它总结了用于编码抗体残基和CDR残基鉴定的概念和策略。最后,它讨论了特定残基组在免疫球蛋白的结合亲和力和/或特异性中的重要性。
关键词: numbering、CDR、 import residues
一、前言
在1986年,Muromonab-CD3是第一种被批准用作人类治疗药物的单克隆抗体(mAb)。 这种针对T淋巴细胞CD3复合物的鼠抗体已被广泛用于预防器官移植患者的急性排斥反应。 迄今为止,美国食品和药物管理局(FDA)批准了71例mAbs1。 这些抗体主要用于治疗癌症和免疫疾病。 此外,多种mAb被证明在治疗各种病症如骨丢失(Denosumab),高胆固醇血症(Evolocumab)或传染病(Raxibacumab,Palivizumab)方面是有效的。
在过去十年中,随着深度测序技术的出现,已经报道了大量新的抗体序列。此外,已经报道了许多与其靶抗原复合的抗体的3D结构,并且已经统计鉴定与抗原直接接触或影响结合亲和力的残基。这有望揭示抗原 - 抗体相互作用的分子基础。尽管已经开发了基于结构数据的不同生物信息学工具来预测抗原表位或已知抗体与其抗原之间的相互作用表面(对接,docking),但计算机方法目前还无法从头定制抗体与靶抗原的特异性。相反,使用体内免疫技术或通过噬菌体展示从组合文库中选择抗体,并且证明其有效获得针对给定目标抗原的特异性抗体。
已充分证明,免疫球蛋白的结构形成Y形糖蛋白(~150kDa),其由两条相同的重链和两条相同的轻链组成。这些重链和轻链各自 由与同一祖先基因分开的基因编码。轻链和重链的可变结构域负责抗原结合,而恒定结构域与免疫系统的其他组分连通。值得注意的是,除了这些“标准”免疫球蛋白外,骆驼科植物以及一些软骨鱼类还表达另一种缺乏轻链的抗体,分别称为重链抗体(HcAb)或免疫球蛋白新抗原受体(IgNAR, immunoglobulin new antigen receptor )。这些同型二聚体抗体能够以与常规异四聚体抗体相似的亲和力结合其抗原。在所有情况下,每条链的可变结构域含有三个称为互补决定区( complementary determining region, CDR-1,-2和-3)的高变环。 CDR以外的结构保守区为框架区(FR-1,-2,-3和-4),形成“核心”β-折叠结构。 CDR序列的长度和组成是高度可变的,尤其是在CDR3中。这种多样性的起源在于遗传机制的复杂性,该遗传机制从相对少量的抗体基因产生高度可变的抗体库。根据V(D)J重组机制,可变区由两个基因(V和J,对于λ和κ轻链)或三个基因(对于重链的V,D和J)组装。连接区域是CDR3的一部分。通过允许在那些连接中添加或缺失核苷酸的机制以及通过重组基因中的体细胞超突变引入CDR3长度和序列的进一步变异性。 CDR通常近似于与抗原相互作用的抗体的paratope,因其含有抗原结合残基。这篇综述将证明,这种paratope的定义过于简单化,并不完全符合现实。
抗体工程方法引起了许多研究团体和制药公司的关注。 抗体工程技术在药物开发中具有越来越重要的意义,并且已经开发了不同的生物治疗剂,包括利用抗体片段,双特异性抗体或抗体 - 药物偶联物。 然而,尽管它们在药物治疗中取得了越来越大的成功,但当注射到患者体内时,mAb通常会引发不良事件,特别是含有鼠或大鼠序列的嵌合分子,并且可能导致患者血清中出现人类抗球蛋白抗体。
为了克服这些问题,已经开发了不同的策略,其成功地降低了mAb的免疫原性并因此降低了免疫不良事件的风险。使mAb人源化的最简单方法在于用动物来源的IgG恒定区替换人免疫球蛋白的相应恒定区。这些所谓的嵌合抗体仍包括来自动物来源的负责抗原结合的完整可变区。然而,在大多数情况下,这些可变区含有足以引发副作用的免疫原性区域,包括过敏反应。因此,开发了可变区的进一步人源化方法。在这种情况下,CDR-移植或特异性确定残基(Specificity Determining Residue,SDR)-移植已成为本领域广泛使用的方法。简而言之,这些方法包括用来自人来源的同源区替换鼠框架区。与嵌合抗体相比,这些所谓的“重构的mAb”显示出更少的免疫原性表位。然而,为了达到更高程度的抗体人源化,需要完整且精确地鉴定免疫原性表位。在这种情况下,各种方法在过去几年中得到了显着改善,并且已经大大改进了人源化方法。这些方法包括表面整形或贴面(surface reshaping or veneering ),超人源化(superhumanization),人体串含量优化(human string content optimization)或使用噬菌体展示文库的组合方法。
不幸的是,即使这些人源化技术产生具有降低的免疫原性的mAb,它们也经常导致抗体亲和力和/或特异性的丧失。 在大多数情况下,这种亲和力丧失的主要原因归因于各种因素,例如CDR序列的不精确定义,用于环嫁接的人框架支架的不适当选择以及错误鉴定来自不同物种的结构相应残基。 实际上,抗体工程技术需要准确鉴定CDR,抗原结合残基以及结构相应的残基。 因此,适当的标准化编号方案至关重要。 不幸的是,建立强大的种间编号公约极具挑战性,特别是鉴于CDR长度和序列的高度可变性。
本综述分为两部分。 第一部分描述和讨论了迄今为止建立的可变区域的不同编号方案。 第二部分比较了不同的CDR定义,并讨论了参与抗原结合的不同残基以及间接影响免疫球蛋白结合亲和力的许多框架残基。最后,我们讨论并提出抗体人源化的一般方法。 足够的抗体编号和注释在抗体工程领域中至关重要,它将有力地推进基于单克隆抗体的人类药物开发。
二、抗体可变域的编号方案 ( NUMBERING SCHEMES OF ANTIBODY VARIABLE DOMAINS )
抗体工程方法需要精确鉴定抗体中对其靶抗原的相互作用和/或亲和力有影响的残基。例如,如上所述,CDR-移植旨在通过改造针对靶抗原的可变区来降低非人抗体的免疫原性。该方法需要准确鉴定CDR,因此需要来自人和非人物种的抗体序列的充分比对。此外,如本综述后面所述,已显示来自框架区的残基也可能对抗体亲和力产生强烈影响。因此,精确鉴定人和动物免疫球蛋白链中的相应位置是必要的。然而,文献中目前可用的不同氨基酸编号方案的使用令人困惑并且可能导致框架和CDR残基的异常鉴定。因此,了解不同的编号方案至关重要,因此能够对它们进行比较。以下部分专门用于描述表S1中比较和总结的不同编号方案。
2.1 Kabat编号方案
在过去的几十年中,抗体的测序和结晶导致各种序列和结构数据库的显着增加,这使得可以比较来自人和动物免疫球蛋白的可变区。 1970年,Kabat和Wu对77个Bence-Jones蛋白和免疫球蛋白轻链序列进行比对,以研究可变抗体区域连续位置的氨基酸组成的统计变异性。他们将“变异性参数”(“variability parameter”)定义为给定位置的不同氨基酸数除以该位置最多氨基酸的频率。该分析揭示了轻链可变区中的三个高变区。还证实了高度保守残基的存在,例如在免疫球蛋白结构域的内核处形成二硫键的两个半胱氨酸和在CDRL1之后立即定位的色氨酸残基。同样,在可变重链结构域中也鉴定了三个相应的高变区。 Kabat和Wu假定这些高变区将聚集在折叠结构域的一侧以形成负责特异性抗原识别的表面,并将这些高变区称为“互补决定区”(Complementarity Determining Regions)“CDR-1,-2和-3” 。该假设后来得到证实并进一步研究以区分这些CDR内的抗原接触或构象重要残基。
1979年,Kabat等人是第一个为免疫球蛋白可变区提出标准化编号方案的人。在他们的“免疫学蛋白质序列”(Sequences of Proteins of Immunological Interest)的汇编中,轻链(λ,κ)可变区和抗体重链的氨基酸序列,以及T细胞受体的可变区(α,β, γ,δ)对齐并编号。他们观察到:
- 分析的序列表现出可变的长度,并且间隙和插入只能包括在精确的位置。
- 有趣的是,插入点位于CDR内,CDRL2除外,也位于框架区内的某些位置。在编号方案中,识别这些插入并用字母注释(例如,27a,27b …)。
- 还值得注意的是,在所有λ轻链中不存在残基L10(真的是这样么??),而λ和κ链由位于不同染色体上的两个不同基因编码。
在过去的几十年中,序列的积累导致了KABATMAN数据库的创建
尽管Kabat编号方案通常被认为是编号抗体残基广泛采用的标准,但它具有一些重要的局限性。
- 首先,该方案建立在来自具有最常见序列长度的抗体的有限数量序列的比对上。因此,不包括在CDR或框架区中具有非常规插入或缺失的序列。因此,最初的Kabat方案忽略了非常规长度的抗体链,具有独特的插入或缺失。然而,根据一个更大的,并定期更新数据库(Abysis3 ),编号可变结构域的氨基酸序列命名ABnum2 一个有用的编号工具,考虑到不同的长度,特别是在CDR2的插入通过在位置L54添加一个插入点。
- Kabat方案的第二个主要限制是它与抗体的3D结构不匹配。实际上,由Kabat定义的高变区与结构抗原结合环不完全匹配。 CDR-L1(L27)和CDR-H1(H35)中定义的插入点与它们在结构中的相应位置不一致(图1)。换句话说,CDR-L1和CDR-H1中晶体结构中的相应残基(拓扑排列)在Kabat编号方案中不共享相同的编号。

图1 | CDRL1氨基酸插入位置根据Kabat和Chothia编号方案,用于蛋白质数据库(PDB)结构1igm和2imm的轻链可变结构域。 (A)根据Kabat或Chothia编号方案的1igm和2imm CDR1L的氨基酸序列。根据Kabat的编号,从较长的环中插入氨基酸置于L27(品红色)后面并用'27a至27f’表示;而根据Chothia的编号方案,他们被置于L30(蓝色)后面。结构上对齐的位置L31以红色显示。 (B)从氨基酸L24到L34的1个和2个月的CDRL1环的叠加的带状表示。颜色代码如(A)中所示,根据Kabat编号,CDRL1(L27a至L27f)中2mm的额外氨基酸以黄色显示。 (C)与(B)中相同的结构重叠,除了黄色补充氨基酸和蓝色氨基酸位置是根据Chothia的编号方案。
简单说明一下: B和C中黄色部分分别代表Kabat和Chothia定义的插入片段,很明显呀,Chothia定义的插入片段在结构上更符合一些呢。
2.2 Chothia编号方案
1987年,Chothia和Lesk为抗体可变区引入了基于结构的编号方案。 它们对齐抗体可变区的晶体结构,定义了形成CDR的环结构,并校正了CDRL1和CDRH1内插入点的位置编号,使它们更适合其拓扑位置(图1)。 此外,他们将重链和轻链的CDR环分类为少量保守结构,称为“规范”(canonical )类,将在后面讨论。
基于抗体结构的比对,Chothia编号方案将氨基酸插入点从位置L27转移到L30并从位置H35转移到H31。 值得一提的是,Chothia CDR定义确保了与结构循环的更好对应。 由Chothia鉴定的CDRH3的环结构与Kabat高变区匹配良好。 相反,除了从H26延伸到H32的CDRH1之外,其他环比Kabat定义的高变序列短。 在任何情况下,根据Kabat在高变氨基酸上定义的,并且基于Chothia命名法中的环拓扑结构的CDR,对于一些CDR具有移位位置和/或包含偏离环长度(图2)。
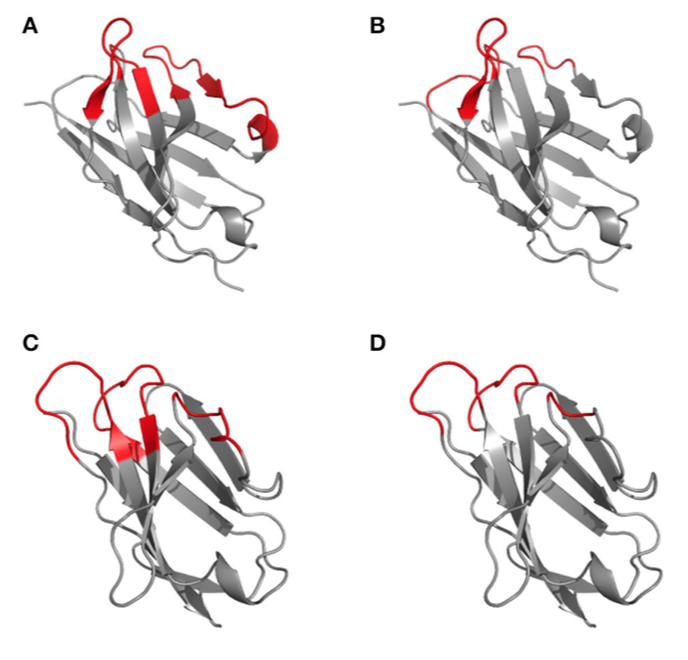
图2 | 根据1kiq结构域结构上显示的Kabat(高变区)(A,C)和Chothia(结构环)(B,D)的CDR定义的差异。 光(A,B)和重(C,D)链的可变域3D结构用卡通表示,框架为灰色,CDR根据Kabat(A,C)和Chothia(B,D)以红色突出显示 编号方案。
Chothia编号方案具有以下主要优点:
- 来自不同抗体的拓扑排列的残基位于相同的位置编号,并且Chothia CDR定义在大多数抗体序列中对应于结构抗原结合环。
然而,与Kabat或IMGT编号方案(见下文)相比,这种编号方案的使用有限,也会产生混淆。此外,后来的研究由Chothia等人发表。将CDR L1中的插入点从残基L30改变为L31。然而,在研究抗原结合环的构象时,在大型数据库中存在的抗体,他们在1997年回到了最初的L30位置。以类似的方式,它们最初在λ轻链中的位置L93处定义了插入点,其在随后的研究中移位到位置L95。
该编号方案的一个重要限制:
- 是由于使用最常见的CDR序列长度,如Kabat编号方案,因此Chothia方案忽略了具有非常规长度的序列。然而,类似于Kabat编号方案,可以通过定义新的插入点来优化该系统。
2.3 Martin编号方案( Martin Numbering Scheme )
在2008年发表的一项研究中,Martin等人重点关注非常规长度的不同框架区域的结构对齐:
- 他们强调了大多数序列和结构中不存在的残基,因此作者将这些定义为缺失位置。
- 通过分析序列和结构,他们还提出了从位置H82到H72的重链结构域的框架区3内的插入点的校正。
- 此外,通过类比CDRH2,他们修改了现在位于L52位置的CDRL2插入点的位置。
- 最后,他们使用了前面提到的编号软件ABnum,并推荐了一种新的编号方案,该方案由ABnum软件纠正的Chothia编号系统组成。实际上,该软件使用更大的Abysis数据库,该数据库集成了来自Kabat,IMGT和PDB数据库的序列。因此,ABnum程序在Chothia和Kabat编号方案中的H6位置定义了一个新的插入/删除位置。 Martin编号方案校正了这个插入点并将其移向位置H8。
Martin编号方案应被视为Chothia编号的最新版本。 通过分析较大数据库中的序列和结构,它可以校正插入位置,定义新的位置并突出显示缺失的位置,以便更好地拟合残基的拓扑位置。
(这应该就是增强版的chothia了吧)
2.4 Gelfand编号方案
1995年,在一些研究中使用的另一种有趣的编号方法是Gelfand等人描述的方法。该编号系统是一种相对复杂的命名。可变链序列被分成称为“单词”(word)的21个片段,这些“单词”中的每一个与二级结构元素(链或环 a strand or a loop)匹配。链由字母顺序的字母(例如,A,B,C)定义,并且环由两个字母定义,其对应于相邻的链(例如,AB,BC …)。然而,这个术语有两个例外:可变链的三个N末端残基(因为它们不是第一个β链的一部分而命名为OA)和连接B和C链的环,其具有’两个跨度桥构造,一个残留物深深地插入结构中(图3)。该循环分为两个单词BC和CB。该编号系统不包括间隙或缺失点,但允许精确比较对齐序列之间的二级结构(环和链)。同样值得注意的是,几个loops的Gelfand定义并不完全对应于Chothia对loops的定义。
(到底谁更准确呢?)
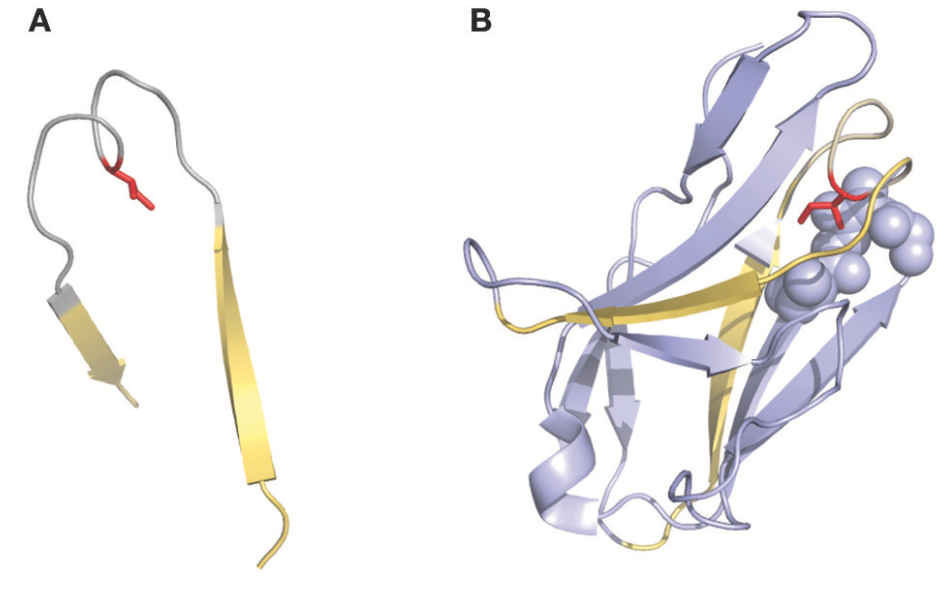
图3 | 表示存在于2fb4抗体结构中的CDRL1的“双跨桥”(two-span-bridge)构象。 双跨桥以灰色表示,边界线以黄色(A,B)显示。 (A)显示带有Ile侧链(红色)的隔离环,指向环结构。 (B)显示Ile如何深入渗透到轻链可变结构域的核心中的完整结构域,其与在口袋(紫色球体)中组织的相邻残基形成疏水相互作用。
2.5 IMGT编号方案
1997年,Lefranc等人为免疫球蛋白超家族的所有蛋白质序列引入了新的标准化编号系统,包括来自抗体轻链和重链的可变结构域以及来自不同物种的T细胞受体链。 它们的编号方案基于种系V基因的氨基酸序列比对。 因此,氨基酸序列和编号在CDR3应该开始的地方停止。 后来,作者将编号方案扩展到整个可变域,并开发了各种工具来分析全长序列。 IMGT拥有自己对框架区域(称为FR-IMGT)和CDR(称为CDR-IMGT)的定义。
IMGT编号方法基于种系V序列(germ-line V)比对从1到128连续计数残基。 因此,它避免使用插入码,除了位置111和112之间的CDR3-IMGT具有超过13个氨基酸。 相反,当特定序列中缺少残基时,不会归因于数字。 例如,在6个氨基酸长的CDR1-IMGT中,残基#27之后是残基#34(并且残基编号#28-#33不存在)。 根据Kabat,Chothia和IMGT编号方案的对齐示例如图4所示。
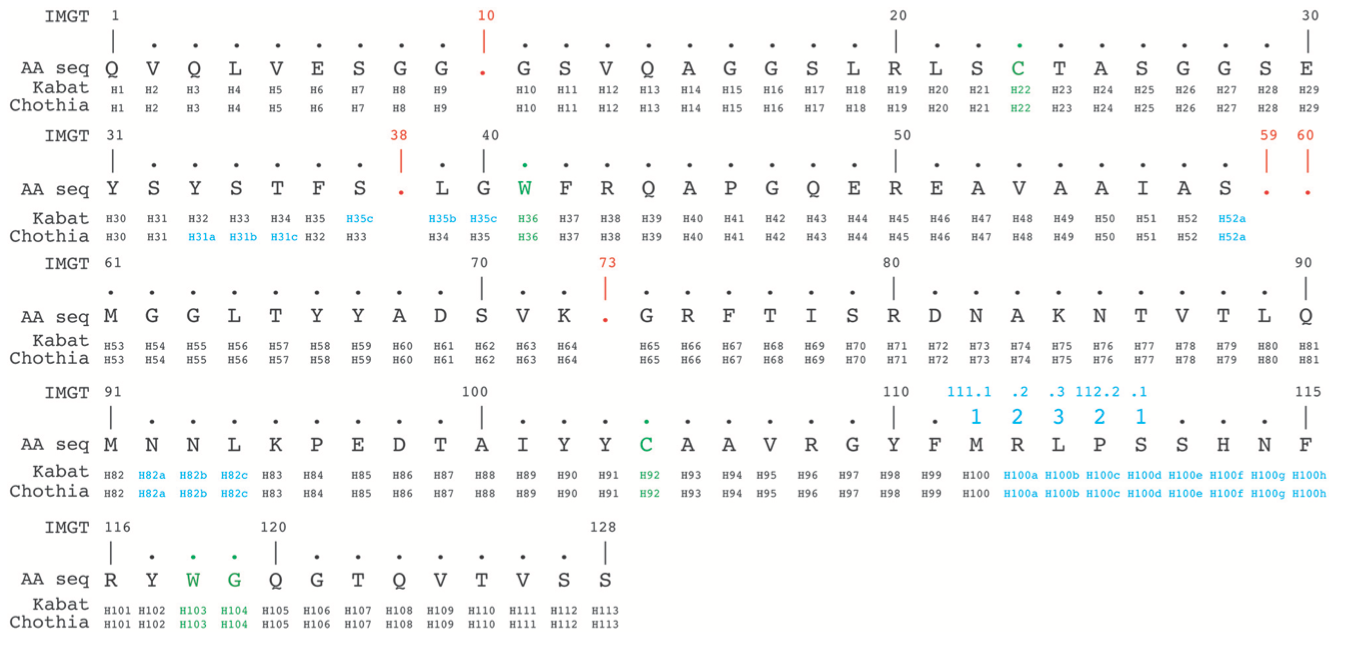
图4 | 根据Kabat,Chothia和IMGT编号方案,从PDB(3dwt)对齐nanobody序列。 在绿色中,半胱氨酸形成保守的二硫键,对抗保守的色氨酸。 此外,CDR3H下游的保守色氨酸118-甘氨酸119(IMGT编号,也是绿色)划分了框架-4区域的起始。 如果在给定位置没有残留物,则IMGT编号方案使用gaps(以红色表示)。 插入位置以蓝色表示。
IMGT是免疫遗传学和免疫信息学的主要参考。 其公约,包括其氨基酸编号方法,已得到世界卫生组织 - 国际免疫学会联合会的认可和使用。 该编号方法的主要优点在于它基于来自包括完整免疫球蛋白超家族的完整参考基因数据库的序列的比对。 这导致了非常有用的工具的开发。 例如,氨基酸比对和编号可以通过IMGT / DomainGapAlign进行。 该工具还能够通过鉴定编码可变区的相应VDJ基因来分析序列结构域多态性。 它与另一个有趣的应用程序IMGT-“Collier de Perles”相结合,可以一目了然地显示二维表示中氨基酸的位置,还可以轻松描绘FR-IMGT和CDR-IMGT。
缺点:
- 然而,由于沿着序列连续编号氨基酸,IMGT编号方案不允许直观地显示插入位置,即使对于最常见的插入位置也是如此。 出于同样的原因,这种编号方案不太灵活。 实际上,在Kabat和Chothia编号系统中,氨基酸插入点的位置很容易合并; 对于具有新氨基酸插入的潜在序列,更难以使IMGT方案适应。
- 必须注意的是,IMGT将所有这些插入置于CDR的末端,这与抗体结构无关。 但是,此问题已在后来的V-Quest软件中得到纠正,该软件将插入放置在CDR-IMGT的中间,这与可用的结构数据更好地匹配

Anchor positions are:
- 26 and 39 for the CDR1-IMGT
- 55 and 66 for the CDR2-IMGT
- 104 and 118 for the CDR3-IMGT
锚位置(Anchor positions)是界定CDR-IMGT并属于相邻FR-IMGT的位置。
2.6 Honneger’s 编号方案(AHo’s)
Honegger方案以homogenized形式编号免疫球蛋白超家族的可变结构域。 该系统基于覆盖观察到的长度变化的免疫球蛋白可变区的3D结构的结构比对。 它允许定义结构上保守的Cα位置,因此推导出适当的框架区和CDR长度(图5)
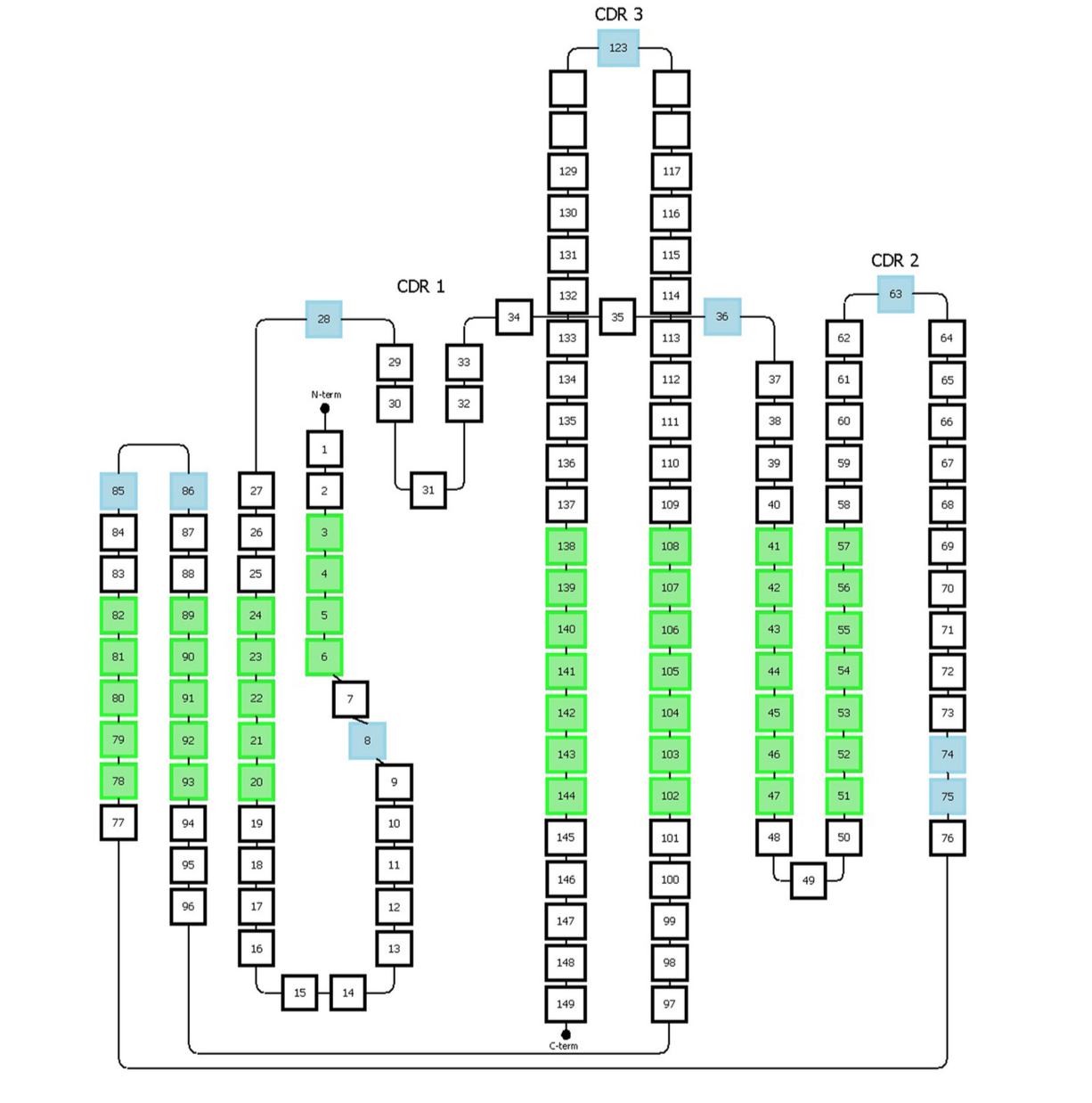
图5 | 代表Honneger编号方案。 容纳间隙的氨基酸位置以蓝色表示。 绿色位置对应于结构上保守的残基,其Cα位置用于结构叠加。 该图已经改编自Honneger等人发表的研究
Honegger编号方案(AHo):
1.定义了保守残基(C23,W43,C106,G140)和特定位置上的缺口(#27-28,#36,#63,#123)。 2. CDR1具有“双跨桥”(“two span bridge” )构象,其由位置#31处的保守疏水残基产生,其深深地插入结构中并因此将环分成两个不同的部分。 3. Honegger方案描述了位于这两个部分上的两个间隙区域,一个位于第一部分(#27和28),另一个位于第二部分(#36)。该惯例考虑了环路两侧存在的插入的可变性。 4. 此外,另外两个插入点分别位于#74和#75位置,以反映T细胞受体α表现出的CDR2环的较短C-末端分支。 5. 额外的间隙位置位于CDR-2和-3环的中间(图5)。 6. 从进一步的结构分析,他们提出将最初位于位置L10的Vκ链中的插入间隙移位到L8。
Honegger编号系统的主要优点是:
- 它基于结构比对,因此它与抗体3D结构特征更好地匹配,其方式与Chothia编号方案类似。
- 此外,正如IMGT计划所述,AHo非常适合于免疫球蛋白超家族中所有蛋白质的编号,通过在CDR 1和2中包含两个gaps。
缺点:
- 但是,与IMGT方案类似,AHo可以跳过一些数字。顺序残差编号,在分析序列编号时可能会令人费解。
- 该编号方案也不太灵活和适应性,包括具有新的或更大插入的免疫球蛋白。尽管涵盖了观察到的长度变化,但是可以通过考虑更多数量的结构来找到新的插入位置/长度。
- 此外,结构最保守的位置仅从28种不同的结构中获得。同样,通过使该方案特异性地适应特定类型免疫球蛋白(例如抗体)的可变区,可以达到更好的定义框架区的精确度。
最后,使用AHo的序列编号可以提交给PyigClassify server。 但是,此服务器似乎没有考虑原始文件中定义的两个插入位置。
三、在CDRS定义中找到你的way
3.1 CDR定义和抗原结合残基
CDR通常被认为是结构化的环(structured loops),其参与抗原结合并表现出超可变氨基酸组成。然而,基于抗体氨基酸序列定义CDR可能是复杂的。实际上,本综述中提出的不同编号方案使用不同的CDR长度定义。此外,如前所示,Kabat(和IMGT)CDR定义基于序列比对,而Chothia CDR定义更好地反映了抗体3D结构中的环结构。在精确定义CDR长度和位置方面缺乏一致性在某种程度上是出乎意料的,因为这些区域很久以前已经显示出对抗原结合活性负责。为了解决CDR定义的差异,一些作者已经考虑了定义CDR序列的不同可能长度。例如,North等人在他们最近对CDR环构象的结构分析中使用更长的序列。
此外,高结合亲和力反映了非常稳定的抗体 - 抗原复合物。这通过paratope和epitope之间的氨基酸残基多个非共价键实现。
- 然而,已经显示CDR内仅20至33%的氨基酸与抗原直接接触。这些残基,名为“特异性决定性残基(Specificity Determining Residues, SDR)”,由Padlan等人首次描述。他们的结果表明这些SDR参与了与抗原的相互作用,并且在大多数情况下,与CDRs最可变的位置相匹配。
- 使用此SDR概念,MacCallum及其同事提出了一种定义CDR的新方法,并重新命名了SDR“接触残留物”(contact residues)。他们还提出接触残基通常位于互补位的中心,正如Chothia之前提到的,非接触残基在塑造CDR环的构象中起作用,因此最佳地定向接触残基以获得有效和特异性的抗原捆绑。
- 最后,他们的研究表明,小抗原倾向于结合具有更凹的界面的互补位,而长凸形的互补位更适合结合酶活性位点并阻断其催化活性。在这种情况下,使用表现出延长长度的CDR3的纳米抗体或牛抗体是特别令人感兴趣的。
同样,Ofran等人使用多结构比对方法来鉴定可变区的抗原结合残基:
- 他们揭示抗原结合残基显示出特定的氨基酸组成,如先前其他组所建议的。
- 特别地,色氨酸和酪氨酸残基高度过表达,而几乎所有其他残基在所有CDR中代表性不足。
- 此外,他们表明,传统的经典CDR鉴定(Kabat,Chothia,IMGT)可能错过约20%的抗原结合残基,并提出了一种鉴定含有这些抗原结合残基的区域的新方法。他们将这些区域命名为Antigen Binding Regions(ABRs),可以使用Paratome5在线工具进行识别。该服务器通过将抗体序列与一组抗体 - 抗原结构复合物进行比较来鉴定ABR。另一个有用的替代工具是proABC( http://circe.med.uniroma1.it/proABC/)。该软件估计每个残基与抗原形成相互作用的概率
Robin等人提出了另一种有趣的方法,使用计算丙氨酸扫描方法研究抗体 - 抗原复合物的结合自由能。他们表明:
- 研究的复合物中80%的结合自由能聚集在非常有限数量的相互作用残基上(4到13之间)。
- 他们强调了30个对结合自由能有重要贡献的位置,其中27个位于CDR内(使用Kabat定义),其余3个位于框架区。
- 所有这些位置都受限于一组氨基酸(Y,G,S,W,D,N),其中芳香族残基是结合自由能的主要贡献者。
- 基于这些残基的身份,可以预测抗原 - 抗体相互作用的性质,并将在稍后讨论(参见讨论部分)。
- 在他们的分析中,CDRH2和CDRH3包括大多数这些残基,而CDRL2在超过一半的研究复合物中根本没有贡献。
- 最后,他们表明Ab-Ag复合物形成涉及3至6个CDR。他们还使用案例研究的实验数据证实了他们的计算分析
有趣的是,Martin已经定义了一套简单的Kabat和Chothia CDR识别规则,并在ANTICALIGN软件中实现。
图6说明了CDR定义的差异。该比较显示,用经典CDR定义(Kabat,Chothia,IMGT)定义的CDR应仅被视为互补位的近似值。根据上述所有概念和观察结果,如果出现以下情况,则应将残留物视为互补位的一部分:
- 和/或与抗原紧密接触;
- 显着且特异地促成抗体 - 抗原复合物形成时发生的阴性吉布斯能量变化。
此外,应该注意:
- 一些接触残基可能对结合自由能的贡献最小,甚至不利于复合物的形成;
- 对于与同源抗原结合非常重要的残基可能对同源抗原和非同源抗原之间的亲和力差异不重要,
- 对于抗原识别至关重要的残基对于结合自由能可能不重要。实际上,互补位及其相应的表位具有形状和化学互补表面。该形状互补性主要涉及建立范德华(π-π)或其他疏水相互作用的芳族残基(W,F,Y)。相反,具有低解离速率常数的稳定的Ag-Ab复合物主要归因于静电相互作用或涉及带电或极性侧链残基的氢键。重要的是,CDR中包含的许多残基不直接与抗原接触,但通过维持附近相互作用残基的最佳构象和方向,也可能在Ag-Ab相互作用中发挥重要作用。
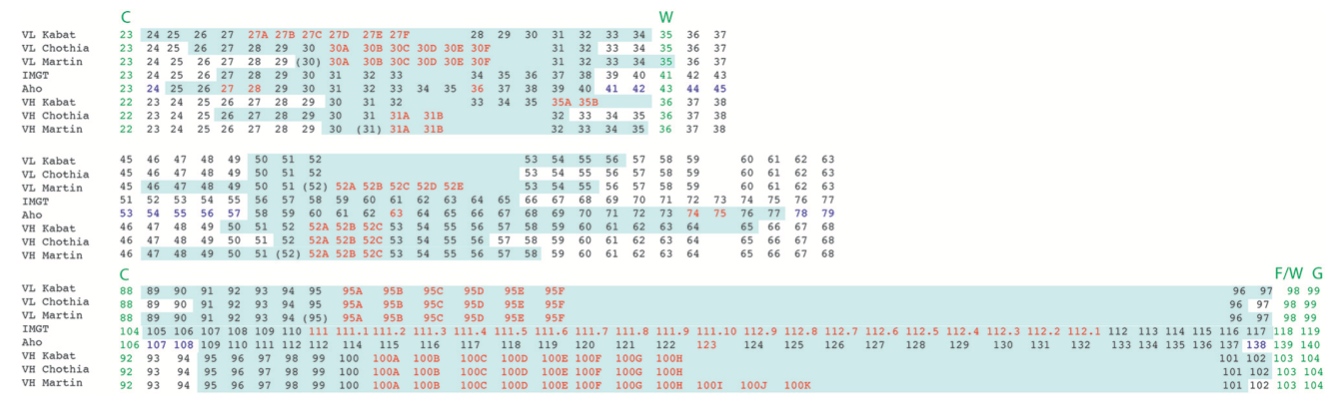
图6 | 根据不同编号方案,轻链和重链可变结构域内CDR的残基位置的比对显示CDR定义中的差异。 CDR以浅绿色突出显示。 氨基酸插入位置以红色表示。 在Aho编号系统中,结构上保守的位置用紫色着色。 对于Martin的CDR定义,CDR对应于抗原接触残基。 CDR1和3周围的保守残基以绿色表示。
(注:跟MOE和 http://www.abysis.org/ ,比较以后,发现这里的CDR定义有问题,比如Chothia L的CDR区间,后续有待考证)
3.2 CDR 结构分类
理想地,CDR的适当定义应包括形成paratope表面并与抗原epitope相互作用的所有残基。因此,CDR的结构分析是重要的。 Chothia的研究小组将重链和轻链的CDR环分类为少数保守结构,称为“规范”类。该分类系统表明轻链的CDR(CDR L1,L2,L3)和重链的前两个CDR(CDR H1,H2)仅采用几种不同的结构。这些结构似乎受标记位置的序列长度和关键氨基酸的存在的限制。作者发现,在CDR和FR区域内仅发现非常少的保守残基(分别在轻链和重链中为13和7)是CDR构象的原因。然而,有争议的是,似乎CDR的种系(germline)序列与其经典类别之间没有明显的相关性,很可能是因为包括在框架区域中的小序列差异可能影响CDR的构象。除了Chothia的研究之外,其他几项研究已经增强了这些CDR的结构聚类以及影响其构象的关键残基的鉴定,包括高度可变的CDRH3。 De Genst等人发表的一项有趣的研究,说明了CDR的序列和结构之间的关系。简而言之,他们发现两个纳米体(nanobodies)中由相同D基因编码的CDR3环的部分采用相同的结构并靶向抗原上的相同表位。
结合起来,这些研究表明,基于CDR序列的结构预测的CDR的结构分类,可以是抗体工程的非常有用的工具。 一些在线工具可用于此目的( http://dunbrack2.fccc.edu/pyigclassify/ , http://opig.stats.ox.ac.uk/webapps/sabdab- sabpred/WelcomeSAbPred.php )。 然而,重要的是要记住,来自框架区的残基也可以影响CDR构象,如本综述中经常讨论的。
四、非CDR残基对抗体结合亲和力的影响
现在已经很好地证实了非CDRs残基可能通过直接接触抗原,或抗体或其抗原结合环的灵活性,或通过构建CDR环本身,通过影响稳定性,在抗体与其抗原的结合亲和力中发挥重要作用。实际上,来自框架区的残基可以调节CDR的构象,因此影响结合亲和力。定义这些残基并命名为“游标区残基”(Vernier zone residues),并且包括位于CDR环附近的框架区中的氨基酸。最后,影响轻链和重链可变结构域包装和定向(packing and orientation )的非CDR残基对于抗原结合亲和力也是关键的,并且令人惊讶地经常被忽略或忽略。因此,Vernier区的氨基酸是不完整的,应该扩展到包括位于框架中的所有残基,这些残基影响抗体的亲和力。下一段重点介绍位于VH和VL之间界面的残基,这些残基改变了这些结构域的堆积(packing),从而影响了互补位的地形。
Chothia等是第一个描述和分析VL和VH结构域的packing的人。他们强调了该界面中涉及的芳香侧链的存在,但他们的分析仅依赖于当时可用的三种抗体结构。 2010年,Abhinandan和Martin对VH和VL包装角度(packing angles )的多样性进行了更全面的分析,包括567种抗体晶体结构。他们开发了一种方法,根据位于这两个结构域之间界面的特定氨基酸的存在来预测重链和轻链可变结构域的包装/方向( packing/orientation )。实际上,它们定义了位于VH和VL结构域之间界面的结构保守残基的Cα原子。然后使用这些原子拟合两条回归线,一条在轻链上,一条在重链上。填充角度值( packing angle )对应于这两个拟合向量形成的角度(图7),并且从一个抗体到另一个抗体(即-31.0°到-60.8°)显着变化,平均值为-45.6°。他们的结果表明,Chothia的环状残留物对这个角度的影响很小。相比之下,作者确定VH/VL界面上的13个残基会影响这种包装角度。PAPS( Packing Angle 预测服务器)的网络工具可在线获取,用于预测包装角度,并基于序列同源性。
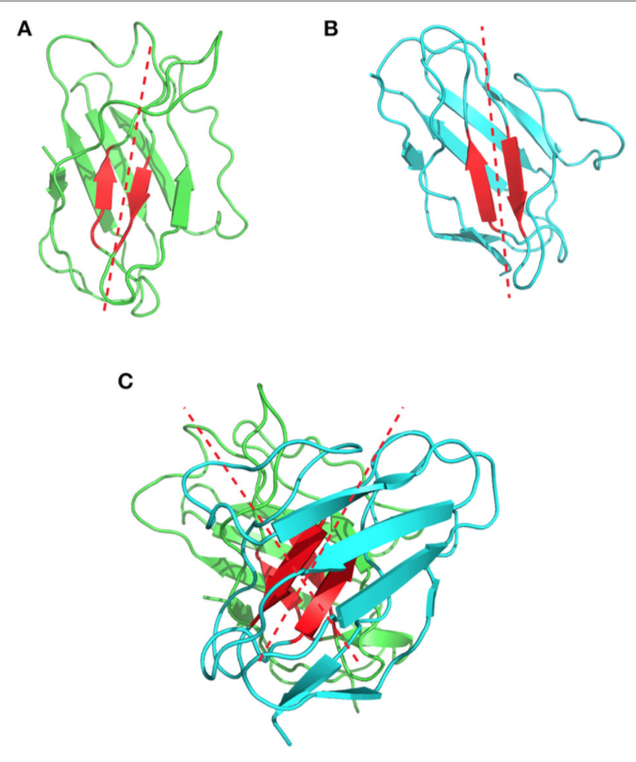
图7 | 表示1bgx结构中的VH / VL填充角。 (A)分别以绿色和青色显示VH和VL的带状结构。 用于定义填充角(packing angle)的保守残留物以红色显示。 拟合在这些残基的Cα原子上的回归线以红色虚线显示。 (B)显示1bgx结构的重链可变结构域,(C)显示1bgx结构的轻链可变结构域。
最近,Dunbar等人进一步表征了可变域的方向。 实际上,一个特定角度可能反映VH和VL的多于一个可能的取向。 因此,他们开发了一种新方法,通过定义5个不同的角度和一个距离来更精确地描述VH / VL方向。 他们还研究了VH / VL界面中的位置以及影响这些不同角度和距离的残基。 他们得出结论,特定位置的特定残基可能导致一个以上的特定VH / VL角。 他们生成了一个名为AB Angle的新软件,该软件可在线获取,用于测量PDB结构的不同角度
该VH / VL角度也影响CDR的相对位置,并因此影响paratope的形状。因此,该参数可以对结合亲和力产生强烈影响。实际上,两个原子之间的结合能是它们遵循Lennard-Jones关系的距离的函数。几个埃的差异可以强烈影响结合自由能的价值。使用直角三角形简单三角法并假设可变区域长度为37埃,VL / VH域之间的1°差异导致CDR表面上暴露的原子位移约0.6。因此,通过CDR-移植技术选择用于人源化的框架区域对于保持亲和力是至关重要的。例如,Nakanishi等表明人源化抗体的严重亲和力丧失,并通过在VH / VL界面进行两次突变恢复了原始亲和力)。同样,Bujotzek等人通过基于预测的VH / VL方向选择人框架进行抗体人源化,并揭示人源化抗体的相似角度和亲和力之间的相关性。
这种包装角度(packing angles)的概念是抗体 - 抗原相互作用的关键方面,因此调节VH / VL方向的残基被认为是在互补位中引入进一步多样性的元件。表S2中列出了显着影响亲和力的框架残基。
五、讨论
5.1 氨基酸编号和CDR定义在抗体人源化中的重要性
为了实现高亲和力结合,paratope及其相应的epitope必须具有大的形状互补表面,此外,接触残基必须建立稳定复合物的相互作用。 影响互补位形状多样性的参数基本上是CDR长度和构象(规范类别,canonical classes),它们的相对取向,结合表面的水合壳(溶剂化)。 两个互补表面的结合主要由建立范德华和疏水相互作用的芳族残基驱动,而复合物的强化涉及相当静电的相互作用和在充分定位的带电荷和极性残基的侧链之间建立的氢键。
在大多数情况下,抗体人源化实验试图通过将非人抗体的CDR移植到人抗体支架来重建原始的互补位-表位相互( paratope-epitope interactions )作用。该CDR移植或再成形方法通常基于抗原 - 抗体相互作用的简化视图,其将互补位减少至抗体的6个CDR。虽然,没有执行抗体人源化的一般方案,因为它总是一个案例到案例的实验,本节试图在这样的练习中提供指导。图8总结并提出了人源化动物来源抗体的标准化方案:
- 首先,我们需要鉴定来自动物来源的供体抗体环内的CDR。在互补位对应于CDR的简化假设下,建议使用Chothia的CDR定义,因为它们与可变区中存在的结构环非常相关。一些在线软件工具可用于CDR结构预测( http://opig.stats.ox.ac.uk/webapps/sabdab- sabpred/WelcomeSAbPred.php , http://rosie.rosettacommons.org/antibody )和分类( http://dunbrack2.fccc.edu/pyigclassify/ )。但是,最好选择最广泛的CDR定义,以确保包含构成互补位的所有残基。
- 其次,正如本综述中反复讨论的那样,经典CDR定义之外的残基也可以是实际互补位的一部分。不同的研究可以帮助识别这些残留物,但不幸的是,目前只有一种生物信息工具(Paratome)可用。
- 最后,CDR的相对取向对于重建互补位表面和充分定位其抗原相互作用残基也是至关重要的。因此,选择最合适的人VH和VL框架支架是至关重要的。选择这种人抗体支架可能与CDR的定义同样重要。与动物抗体中的那些相比,所选择的人框架支架应显示最接近的VH / VL角,以在重构的构建体中正确定位CDR。在这种情况下,突出了它们在VH / VL角度中的作用的不同氨基酸位置,并且目前可获得各种角度预测软件工具。因此,人源化抗体的重塑努力应该是有效的“互补嫁接”(“paratope grafting”)实验,而不是“CDR移植”(“CDR-grafting”)。
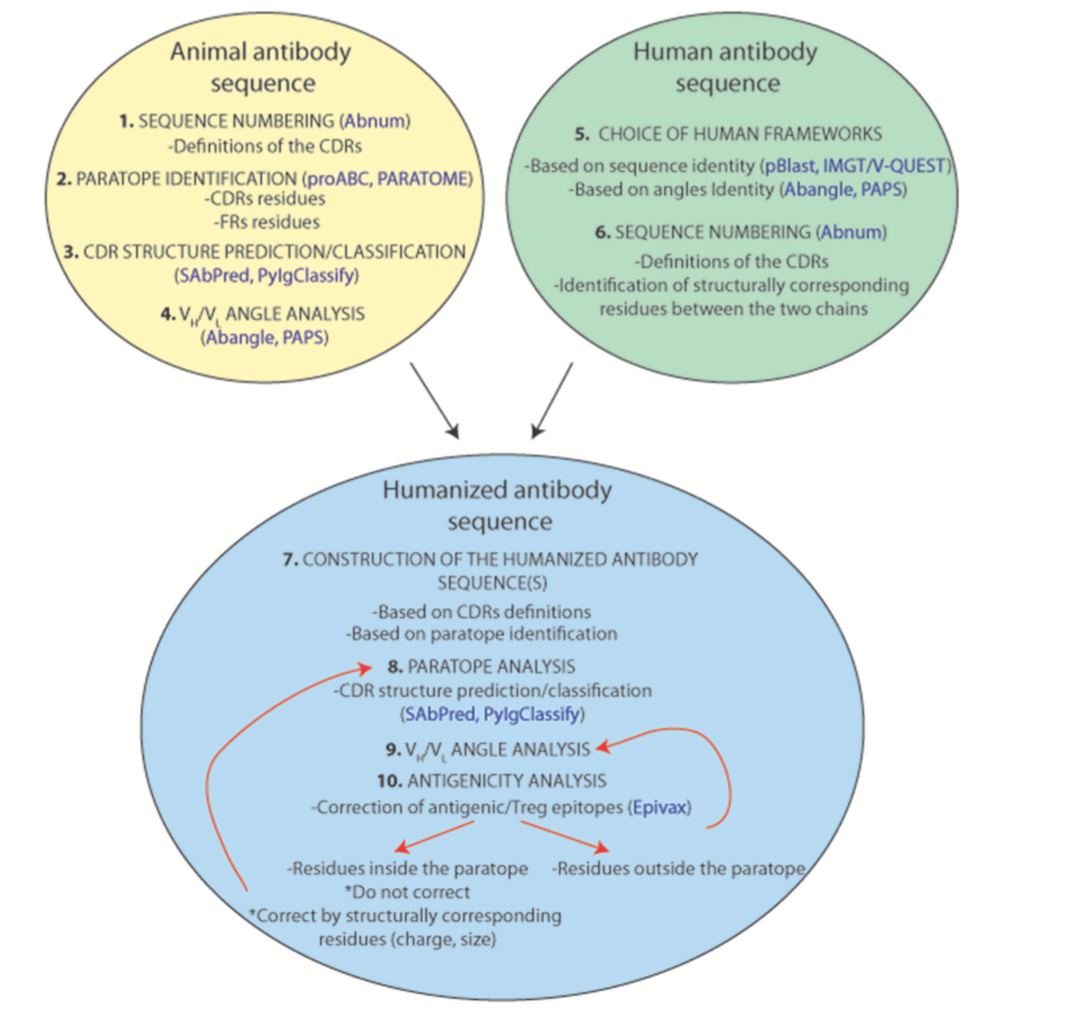
图8 | 表示标准化的人性化流程。 预测工具以蓝色表示。
值得注意的是,出于所有这些目的,重要的是正确对齐抗体序列并精确鉴定具有不同来源链中的叠加位置的残基。 因此,处理相同数量的残基(包括固定的可能残基插入点)以在免疫球蛋白链中占据相同结构位置的编号方案形成了所有抗体工程任务的先决条件。 在这方面,增强的Chothia(马丁)编号系统更容易使用,因为它确定了精确的插入点,但当然,这种选择是非常主观的。
另一个关键点是人源化抗体不应该在患者中诱导任何不利的免疫反应。因此,应该进行蛋白质序列中潜在免疫原性表位的预测。简而言之,这种不利的免疫应答是在抗原呈递细胞内化抗体后发生的。抗体在与HLA-DR,HLA-DQ或HLA-DP分子结合的寡肽中被消化。这些膜蛋白复合物结合特异性寡肽并将它们呈递给激活免疫应答的淋巴细胞T辅助细胞。如今,报告这些MHC-II分子等位基因多态性的数据库可用于预测寡肽结合潜力。其中之一是Epivax网络工具,其包括扫描蛋白质序列以鉴定推定的T细胞表位。预测鉴定的抗原序列与HLA-II DR(或其他HLA-II同种型)蛋白结合,但该程序还可鉴定抑制免疫应答的Treg表位。该软件提供突变一个或多个氨基酸以降低蛋白质序列的免疫原性。计算序列免疫原性并以“Epimatrix评分”表示,以允许预测给定氨基酸序列的免疫原性。突出显示不应突变的重要残基(用于结合亲和力/特异性,角度等)可改善该软件。重要的是要意识到,在抗体的情况下,在维持高亲和力和低免疫原性之间可能发生冲突。这是抗体人源化仍然具有挑战性的原因之一。
六、结论
本综述描述了目前可用的不同氨基酸编号系统和CDR定义,并强调了标准化编号系统对抗体工程策略的重要性,尤其是抗体人源化任务。 实际上,有效的氨基酸编号系统应该能够将相同数量的残基分配给来自不同物种的抗体中的结构上对齐的位置。 尽管基于不断增长的数据库的几种编号工具可在线获得,但建议比较不同的编号系统,因为仍然存在不准确性,特别是对于具有非常规长度的可变抗体结构域。
此外,已经审查了不同的CDR定义和其他概念,例如接触残基或抗原结合残基。在抗体人源化方法的背景下,互补位(paratope)通常限于CDR。这种近似是有用的,只要它允许CDR-移植方法是一种简单且通用的人源化工具。相反,接枝接触残基(grafting contacting residues)(甚至CDR之外的那些)和/或对结合自由能有影响的残基将允许更好地重构互补位。然而,这种方法在抗体人源化工作中不太方便,因为互补位的不连续性质与需要精确测定这些残基的实验方法相结合。此外,已经描述了分析轻链和重链可变区之间的角度的不同研究。在CDR移植或其他抗体人源化方法中,应考虑影响VL / VH包装角度的残基以恢复完全结合亲和力。最后,所有这些对抗体人源化至关重要的概念都应包括在人源化过程中。
总之,使用适当的氨基酸编号方案精确鉴定的互补位对于改造具有高亲和力,稳定性和低免疫原性的人源化抗体是必需的。 在另一个框架上设计一个功能齐全的互补位不仅应限于移植抗原接触和相互作用的残基,还应包括有助于固定抗原结合环的氨基酸和影响配对VL / VH相对方向的位置的残基。所有这些残基必须占据3D结构中的相同位置。
参考资料
- 《Understanding the Significance and Implications of Antibody Numbering and Antigen-Binding Surface/Residue Definition》
- http://www.imgt.org/IMGTScientificChart/Nomenclature/IMGT-FRCDRdefinition.html
- http://bioinf.org.uk/abs/info.html
