【2.4.2】双特异性抗体作用机理
摘要
双特异性抗体(bsAb)用于描述旨在识别两种不同表位或抗原的分子大家族。 BsAb的形式多种多样,从相对较小的蛋白质(仅由两个连接的抗原结合片段组成),到带有附加结构域的大型免疫球蛋白G(IgG)-like分子。有吸引力的bsAb功能是它们具有新颖功能的潜力-也就是说,亲本或参考抗体混合物中不存在的活性。在这些所谓的专一性bsAb中,两种结合特异性的物理联系产生了一种依赖性,该依赖性可以是暂时的,结合事件顺序发生,或者是空间的,结合事件同时发生,例如将效应子连接至靶细胞。迄今为止,已有20多种不同的商业化技术平台可用于bsAb的创建和开发,有2种bsAb已上市,并且有超过85种正在进行临床开发。在这里,我们从机理(mechanistic)的角度回顾了当前的bsAb格局,包括对管线(pipeline)的全面概述。
具有两个不同抗原结合位点的人为抗体基分子的原始概念(双特异性抗体(bsAb))最早是50年前由Nisonoff及其同事提出的,并与对抗体结构的首次见解平行。 他们使用温和的再氧化结合不同特异性的兔抗原结合片段(Fabs),证明了由双特异性片段介导的两种不同细胞类型的凝集。 随后,在产生bsAb方面的连续概念和技术创新与抗体工程和抗体生物学领域的里程碑式发展一起发展(图1),从而导致了当今广泛收集的100多种bsAb形式的收集。 其中约四分之一已开发成技术平台,并已由生物技术公司和制药公司商业化,以产生新颖和差异化的治疗剂。
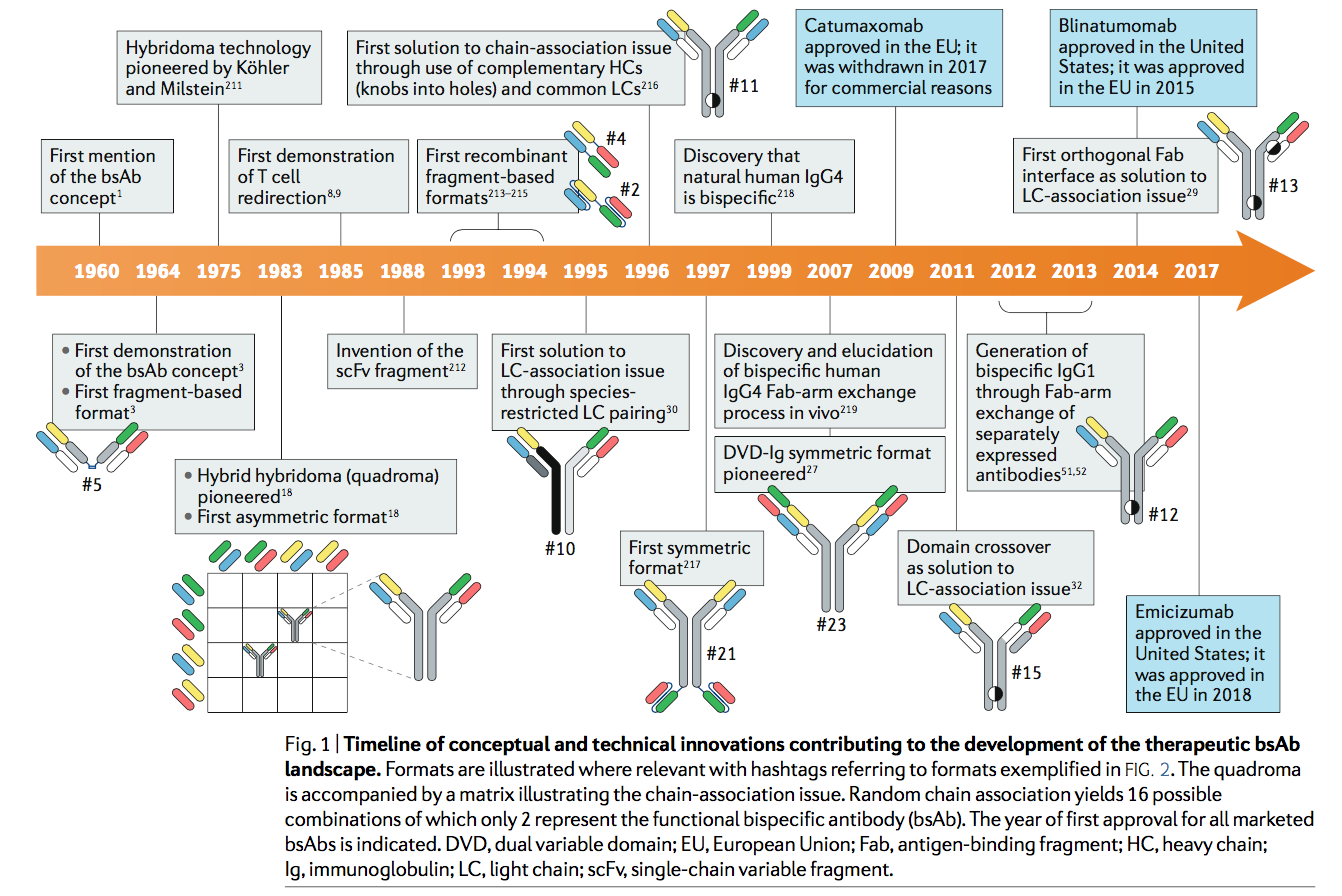
由bsAbs激活的双重靶向概念具有巨大的治疗前景,但是将这些概念转化为治疗已证明具有挑战性。例如,原型bsAb的应用-T细胞的重定向和参与-在1980年代中期首次被描述[8,9],但直到2009年,在经欧盟批准使用catumaxomab进行腹膜内治疗恶性腹水之前,该患者才得以使用。用catumaxomab(T淋巴细胞抗原CD3×上皮细胞粘附分子(EpCAM)bsAb;×表示两种抗原特异性的组合)静脉给药是不可行的,并且在低剂量下会引起致命毒性,这与Fc介导肝内脱靶T细胞活化。尽管由于商业原因最近在2017年将catumaxomab撤出市场,但另一项批准的与T细胞结合的bsAb(T cell-engaging bsAb, bsTCE),blinatumo-mab(CD3×B淋巴细胞抗原CD19)的令人印象深刻的临床结果激发了人们的重新兴趣和投资这个概念。这反映在当前针对血液学和实体瘤适应症的临床开发中的43个T细胞重定向bsAb中。这一成功还刺激了这一概念的进一步发展,并探索了用于T细胞参与的其他触发分子(例如,Vγ9Vδ2T细胞受体(TCR))。此外,正在研究联合疗法,并将其与其他成功的T细胞活化策略(例如检查点抑制剂和嵌合抗原受体(CAR)T细胞方法)进行比较。
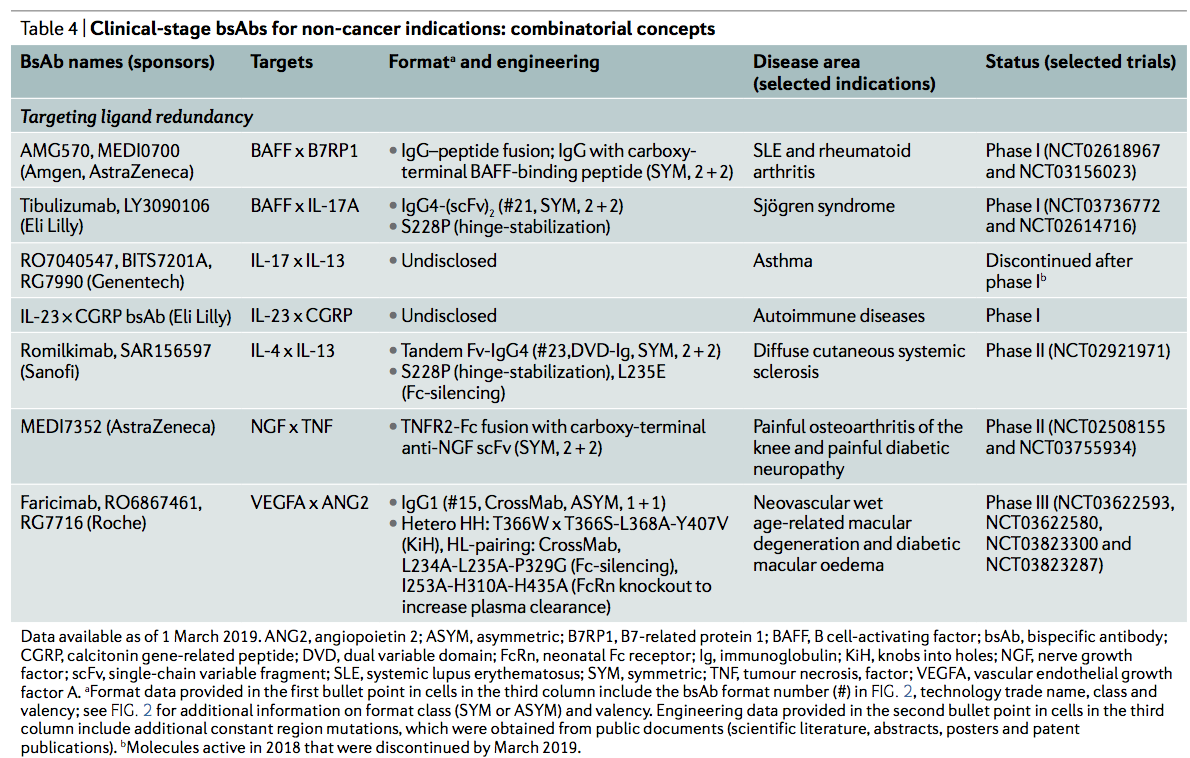
除癌症外,炎症性疾病通常已成为bsAbs临床开发的重点。 然而,在非癌症适应症中bsAb首次进入市场,是在2017年11月美国食品药品监督管理局(FDA)批准艾米珠单抗(凝血因子IXa(FIXa)×FX)用于治疗A15血友病。 此外,还正在探索癌症和炎症性疾病以外的其他疾病领域,包括在I期临床试验中用于糖尿病和HIV感染的临床候选药物,以及在其他各种疾病,包括几种病毒性肝炎和 细菌感染,阿尔茨海默氏病,骨质疏松症和再生医学。 bsAb概念与在这些双重靶向方法中仅混合抗体区别开来的机制令人惊讶地是多种多样的,如以下许多关键示例所述。
例如,通过针对疾病介导的配体和受体中的冗余的组合bsAb或靶向bsAb的串扰信号级联反应,正在开发针对复杂疾病的多因素性质的bsAb。但是,从概念上讲,人们可能会认为组合策略并未利用bsAb可能提供的全部分化。 bsAb的引人注目的特征之一是它们具有显示任何亲本抗体组合中都不存在的活性的潜力(即,产生依赖于两种特异性的物理连接的新功能)。由于这种获得的新活性,尽管我们更喜欢术语专性(term obligate)bsAbs,但这类抗体被称为专性(obligatory)bsAbs。在本综述中,我们将使用机理框架描述bsAb的最新进展和新兴应用,重点是专断(obligate)概念。我们还将讨论将治疗性bsAb概念转化为临床的进展,并提供该领域的未来前景。
-
组合抗体(Combinatorial bsAbs): 显示活性或功能的双特异性抗体(bsAb),也可以通过组合具有相同特异性的单独抗体(例如,亲本或参考抗体混合物)获得。
-
专一性抗体(Obligate bsAbs): 双特异性抗体(bsAbs),其显示的活性或功能取决于两种特异性的物理连接(不能通过将具有相同特异性的单独抗体组合在一起获得)。 这些bsAb介导的双重靶向概念被认为是专一(obligate)的概念。
一、格式 Formats
在天然二价抗体中,两个抗原结合位点是相同的,并且由来自重链(H)和轻链(L)可变域的决定簇组成。因此,bsAb发展的最初挑战之一(通过两个不同的H和两个不同的L链的共表达)是从十种可能的H2L2重组混合物中获得功能性bsAb,通常被称为链缔合问题(Combinatorial bsAbs)【18】。
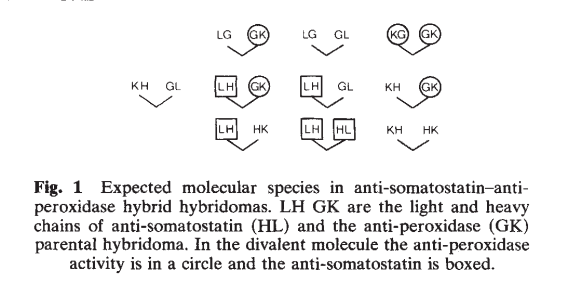
在过去的几十年中,已经开发出许多策略来规避或解决该问题,其特定目的是提高所需最终产品的均质性(homogeneity)和产量。这些策略引入的不同设计特征或功能特性都可以用于对所得bsAb格式的大量集合进行分类。出于本次综述的目的,我们将着重于机制,我们将遵循体系结构分类,并简要讨论机会和局限性。基于结合位点数量(价)的其他区分将进一步细分格式,因为这会影响双重目标和潜在的多重目标应用(例如亲和力或交联诱导的激动作用)(图2)。

1.1 基于片段的格式 (Fragment-based formats)
第一类代表了设计双特异性分子的最小方法,它简单地在一个没有Fc区的分子中结合了多个抗原结合部分(即抗体片段),从而规避了链缔合问题。 这种复杂性的缺乏和(糖基化的)Fc区的缺乏使得在较低的真核和原核表达系统中通过(共)表达1-2条多肽链可以相对简单地产生这些形式,从而具有高产量和降低成本的优势 。
但是,缺乏Fc的形式血浆半衰期相对较短,因为它们缺乏新生Fc受体FcRn对代谢的保护。根据预期的治疗应用,可能需要重新格式化为Fc融合蛋白或引入人血清白蛋白(HSA)靶向部分,以进行临床开发(见下文)。它们缺乏Fc介导的效应子功能或针对特定需要定制此类功能的能力可能代表了另一个缺点。此外,基于片段的产品可能会遇到稳定性和聚集性问题,需要对最终产品进行重新设计。
由于此类的模块化性质,因此可以定制这两种特异性的效价以适合应用,因此可以采用1 +1、1 + 2和2 + 2设计的格式(即,指定用于每种特异性)正在临床中进行评估(图2;请参见下文)。将来可能还会出现其他排列方式,例如1 + 3或3 + 3。
1.2 对称格式 Symmetric formats
在保留Fc区的同时,避免链缔合问题的另一种设计策略是将两种特异性结合在单个多肽链或单个HL对中。所得的形式包括上述基于片段的形式的Fc融合蛋白(以改善药代动力学特性或效应子功能)和将抗体片段与常规抗体分子融合的形式。对称设计允许通过(共)表达1-2条多肽链来产生这些形式。对称格式更类似于天然抗体,但大小和结构不同。这些差异可能会对与天然抗体相关的有利特性(例如稳定性和溶解性)产生负面影响,从而可能损害这些试剂的物理化学和/或药代动力学特性。
由于此类的对称性质,临床开发中的大多数格式都具有四价2 + 2设计。但是,抗原结合位点的紧邻可能会削弱两个靶标的最佳结合,从而潜在地降低功能价,并且可能需要优化(例如,接头长度和结构域位置)各个先导候选物。
1.3 非对称格式
用于生成第三类格式的大多数方法都试图尽可能地保留天然抗体的天然结构,以保留相关的功能特性和良好的质量属性。这表明需要解决链关联问题,并且破坏了H2L2组件的对称性。结果,大多数不对称格式是由于在共表达四个多肽链(或三个,如果是共同的L链或H链是共表达)期间强迫正确的HL链配对和/或促进H链异二聚化的策略产生的。此外,通过基于差异蛋白A结合,顺序亲和色谱法或大小差异设计纯化策略,可以利用不对称性分离所需的最终产物。避免HL链错配的替代策略是将两种特异性分别表达为半分子或亲本抗体,然后分别由促进H链异二聚化的突变驱动的抗体半分子的生产后组装或重组(例如,受控Fab臂交换(cFAE))。
由于大多数不对称形式与天然抗体非常相似,并且缺乏其他非天然抗体结构域或接头序列,因此人们认为它们具有最低的免疫原性。但是,解决链关联问题可能涉及的精心设计的工程可能会抵消其中某些形式的这种优势。此类的不对称性质还可以推断,具有规则免疫球蛋白G(IgG)结构的bsAb通常对每个靶标均具有功能性单价(1 +1)。应当指出,与允许多价靶标结合的格式相比,非对称格式的亲和力降低可能会影响某些应用的效力。
-
连锁关联问题 :两条不同的重链(H)和两条不同的轻链(l)的共表达会产生16种可能的H2l2重组的复杂混合物,代表十种不同的抗体。这些抗体中只有一种(由两种可能的H2l2重组表示)与所需的双特异性抗体相对应(混合物中的最大产率为12.5%)。 通过迫使同源HL配对和/或促进两个 不同H链的异二聚化的策略解决了该问题。
-
价(Valency): 抗体分子中抗原结合位点的数量。 双特异性抗体(bsAb)格式的设计会影响每个靶标的结合位点数量。 每个靶标具有一个结合位点的二价bsAb表示为1 +1。掺入其他结合位点可导致三价(2 +1)和四价(2 + 2或1 + 3)设计。
-
抗体片段 (Antibody fragments): 抗体分子由不同的域组成,可以分别表达并用作模块化构件。 在基于抗体的治疗剂的设计中,涉及抗原重组的结构域通常用作结合部分。 示例包括域抗体(仅重链可变域(VHH))和单链Fv片段(scFvs),抗原结合片段(Fabs),单链Fab片段(scFabs),以及最近的单链Fc 片段(scFc)。
二、bsAb管道的机理
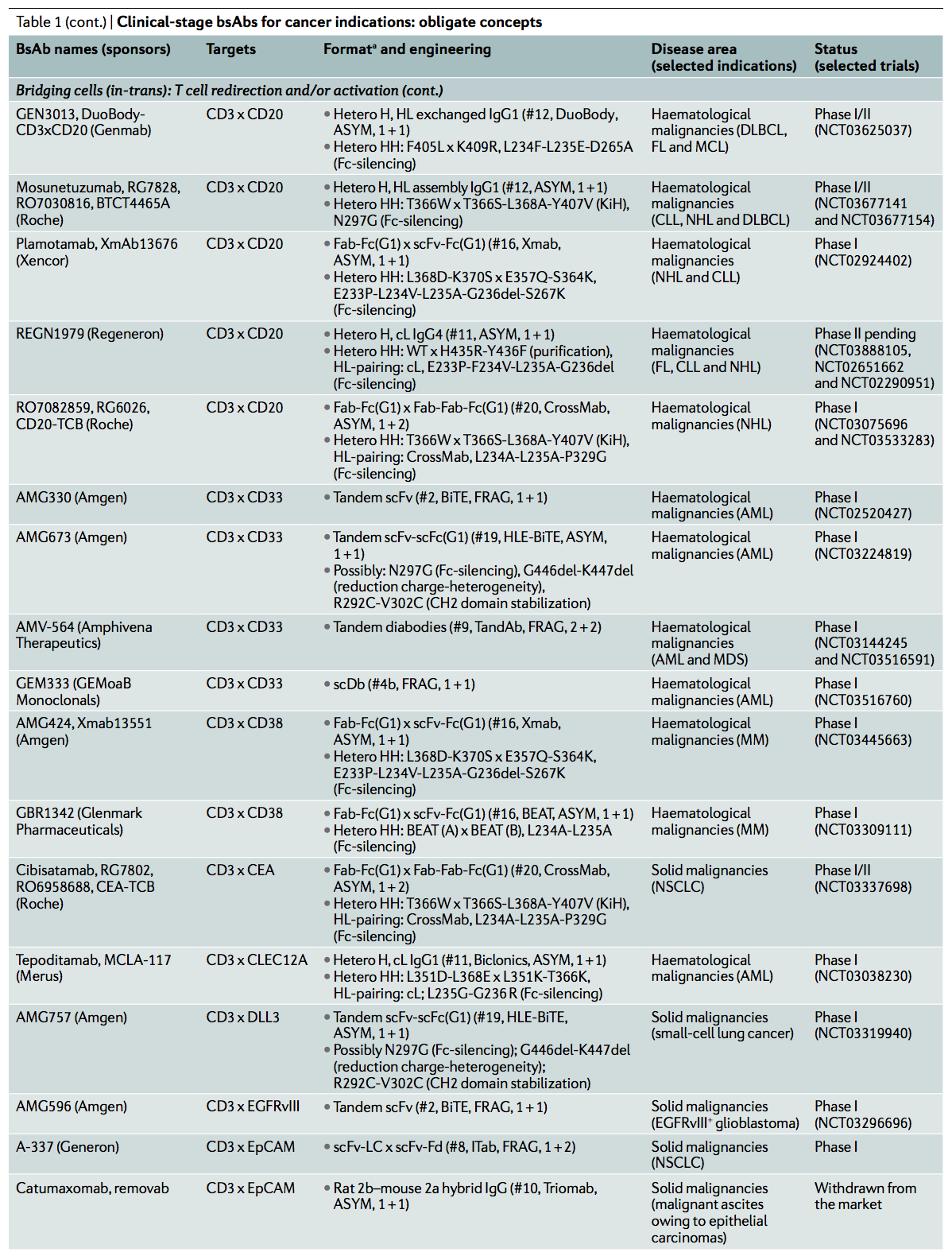
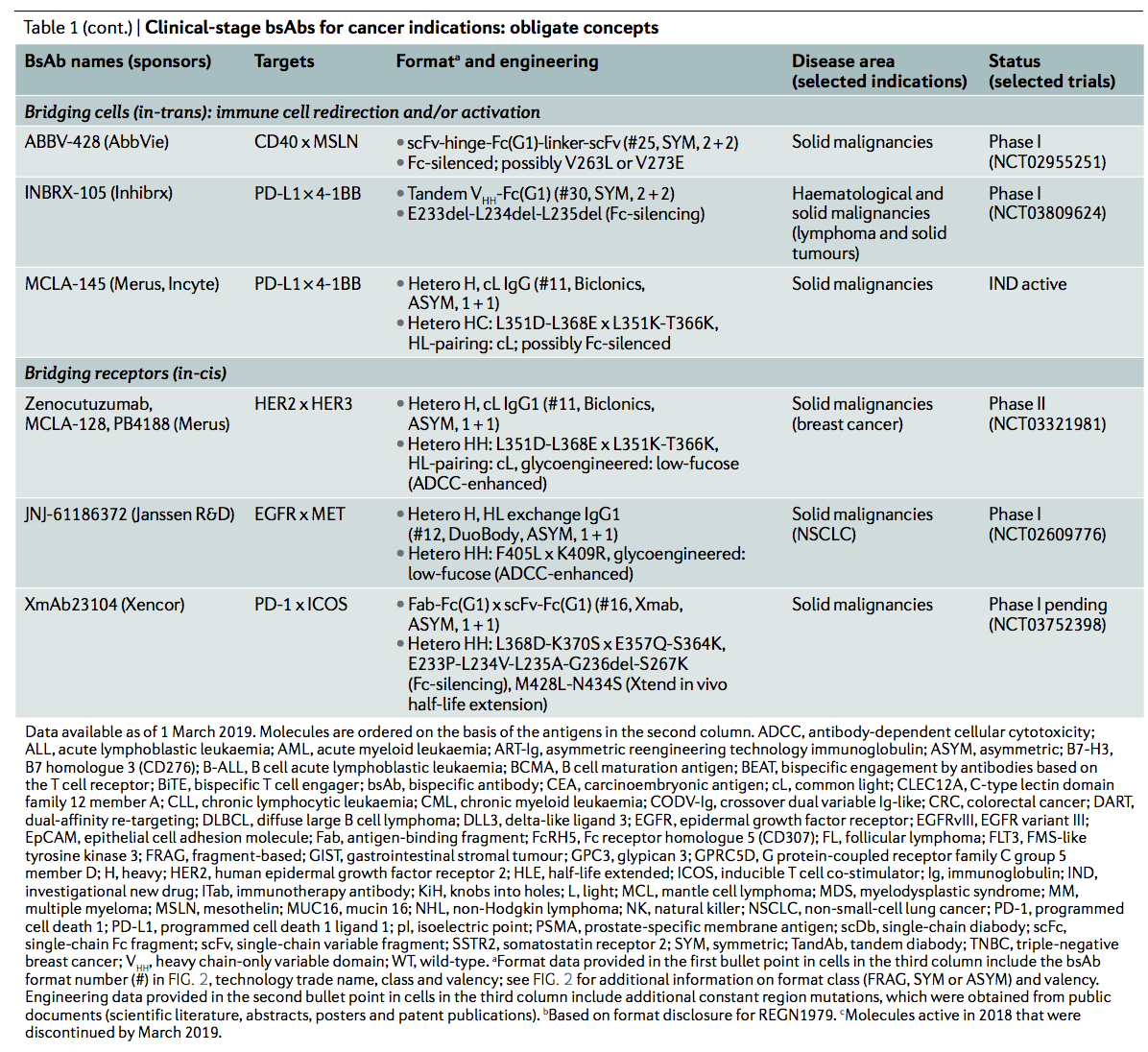
截至2019年3月,商业临床管线包括超过85种bsAb(图3)。反映出近期人们对开发用于癌症治疗的bsAb的浓厚兴趣(图4),在癌症患者中评估了约86%。桥接细胞(bridge cells)作为其专一的作用机制的BsAb代表了最大的群体,其中T细胞重定向是最常见的分母(denominator)(表1,2)。

专性(obligate)bsAbs的优势在于它们能够解锁需要在同一分子中连接两个结合特异性的新功能的能力。可以将其用于创新的治疗概念,例如,桥接两种细胞类型(反式结合)或使两个分子结合在一个细胞膜上(顺式结合)。这些概念要求同时结合两个特异性,而其他专性概念则基于两个结合结构域的顺序结合。专性bsAb概念及其设计和作用机理的例子在图5中进行了总结,并在本节中进行了讨论。
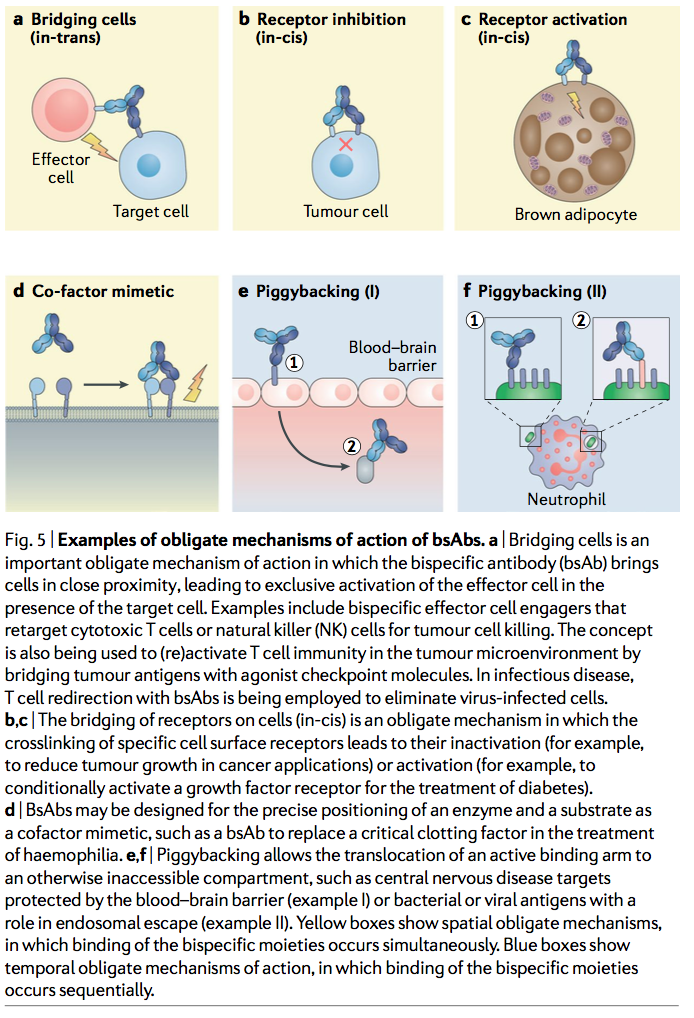
2.1 桥接细胞(反式结合) Bridging cells (in-trans binding)
T细胞重定向:历史的观点 T cell redirection: a historical perspective.
最典型的专一性bsAb概念是将效应T细胞的细胞毒活性重定向至特异性消除肿瘤细胞。 通过这种方法,T细胞通过由T细胞结合域和肿瘤结合域组成的bsAb与肿瘤细胞物理连接(图5a)。 这些bsTCE主要通过TCR复合物中CD3ε的结合来激活T细胞,从而绕过主要的组织相容性复合物(MHC)限制,并引起独立于TCR表位特异性的激活。 尽管概念验证的bsTCEs在共培养试验中显示出很高的效力,但在临床上,这些分子以极低的剂量诱导了T细胞介导的细胞因子快速且不受控制的释放,而未注意到持久的临床活性 。 这些bsAb的产生和稳定性都伴随着并发症,这大大降低了当时进一步开发此类药物的意愿。
但是,当首次出现blinatumomab临床资料时,人们对bsTCE的兴趣恢复了。 Blinatumomab是一种基于抗体片段的小bsAb,分子量约为55 kDa,缺少Fc域,在体内的血浆半衰期很短(1.25±0.63小时)。 当通过持续静脉内输注给予blinatumomab以达到所需的谷值水平时,非霍奇金淋巴瘤(NHL)患者以非常低的剂量观察到了令人印象深刻的反应。 后来,当焦点转移到复发性和/或难治性(r/r)急性淋巴细胞白血病(ALL)时,报告的完全缓解率为43%。 这项研究的结果为2014年FDA批准blinatumomab用于r/r ALL的治疗奠定了基础。
这些令人兴奋的临床数据促使公司寻找有效产生和产生稳定bsAb的解决方案(see Formats)。 结果,可获得大量可开发的bsAb格式,并且许多已进入临床试验。 目前,在临床试验中接受评估的bsAb大约有一半是bsTCE(51%; n = 44/86(图3; 表1))。


bsTCE中的Fc区:活跃,被抑制或不存在。 The Fc region in bsTCEs: active, suppressed or absent.
单特异性鼠抗人CD3抗体OKT3(也称为muromonab-CD3)和开创性的bsTCE catumaxomab的临床经验为安全使用靶向CD3的抗体提供了重要的经验教训。
OKT3已在移植医学中用作免疫抑制剂,但在患者中,特别是在首次给药时,表现出释放的T细胞活化和严重的细胞因子释放。观察到的细胞因子释放的主要机制涉及CD3抗体通过CD3抗体的Fc区与其他免疫细胞上的Fc受体的结合而在T细胞上聚集。为防止CD3不受限制地聚集,开发了具有减少的FcγR结合(和补体因子C1q结合)的第二代人源化CD3抗体(huOKT3γ1(L234A-L235A)),并显示细胞因子释放显着降低。同样,将静脉注射CD3×EpCAM bsTCE catumaxomab引起的严重不良事件归因于其活性Fc区与肝中表达FcγR的Kupffer细胞的脱靶结合。结果,诱导了健壮的局部细胞因子释放和T细胞介导的肝毒性,这对一名患者是致命的。
除了这些安全问题外,我们最近还发现,在体内的同基因模型中,含有具有有效功能的主链的靶向CD3的bsAb的治疗活性也有所降低。这表明Fc介导的效应子机制可能阻碍肿瘤特异性T细胞的重定向和肿瘤细胞的杀伤。
总之,这表明靶向CD3的bsTCEs最好完全抑制Fc介导的效应子功能,以最大程度地降低脱靶毒性,并使治疗功效最大化。实际上,当前在临床中的所有靶向CD3的bsAb都经过工程改造的Fc域以减少FcγR结合,或者是设计上缺少Fc区的双特异性抗体片段。但是,应注意的是,用于抑制FcγR结合的突变在不同形式之间有所不同,某些含Fc的bsTCE比其他形式更惰性。实际上,优选高度惰性的Fc区域,其没有残留的FcγR和C1q相互作用,但其中保留了FcRn结合。
T细胞靶向
尽管已经研究了其他T细胞靶标,例如αβTCR66和T细胞表面糖蛋白CD5来重定向或接合T细胞,但是靶向CD3ε的bsTCE最先进。 迄今为止公开的大多数CD3ε结合单位都来自有限数量的鼠源抗体克隆-通常是非人灵长类交叉反应性单克隆抗体(mAb)SP34-已被人源化,去免疫和/或亲和力成熟 。
几项研究表明,对CD3的亲和力显着影响bsTCE的生物分布。 尽管具有高CD3亲和力的bsTCE(解离常数(KD)<1 nmol / L)在体外共培养试验中显示出优异的效果,但较低的CD3结合臂亲和力(KD =〜50–200 nmol / L)是可取的 ,可以在体内有效分配肿瘤,而无需快速CD3介导的血浆清除或抗体在含T细胞的组织(如脾脏和淋巴结)中捕获。 在这些研究中,需要存在肿瘤特异性臂来驱动肿瘤分布,而在没有肿瘤相关抗原(TAA,tumour-associated antigen)结合臂的情况下,未观察到肿瘤特异性分布。
除亲和力外,CD3结合的价可能会影响分子的活性。虽然二价CD3的结合对于单特异性CD3抗体诱导的抗原调节和耐受性是关键,但在bsTCE的背景下,可能需要使用单价CD3臂。虽然单价结合足以诱导肿瘤特异性T细胞活化,但由于CD3分子在T细胞表面交联,它可以防止抗原调节或细胞因子释放。尽管正在开发的大多数bsTCE确实包含单个CD3结合臂,但一些临床阶段的双特异性分子具有两个CD3结合结构域,但尚不清楚这些格式是否还功能性结合CD3双价。例子包括CD3×CD33(也称为SIGLEC-3)定向的双抗体(TandAb)AMV-564(Amphivena Therapeutics),CD3×CD19 TandAb AFM11(Affimed)和CD3×CD123 Adaptir分子APVO436(Aptevo Therapeutics) 。这些分子显示出有效的T细胞活化,这完全取决于共培养测定中抗原表达细胞的存在。这些bsTCE的安全性和有效性目前正在I期临床试验中进行评估(表1)。
靶向肿瘤细胞
BsTCE直接与T细胞和肿瘤细胞结合形成免疫突触,从而导致TCR活化,颗粒酶和穿孔素的释放,并最终靶向细胞裂解。这基本上类似于TCR肽负载的MHC(pMHC)相互作用介导的裂解突触形成的机制,包括事件序列,分子组成和信号转导。可能的差异是bsTCE可能比天然TCR-pMHC复合物诱导更大的突触数量。这可能是bsTCEs可以建立更多数量的接触点的结果。相比之下,不到十种TCR-pMHC复合物可以诱导T细胞活化。同样,对于高亲和力(〜30–300 pM)的可溶性TCR×CD3双特异性分子,在每个细胞10–150分子的低抗原密度水平下观察到了杀伤力。对于bsTCE,理论上,根据TAA的表达水平,可以建立50至100,000个接触点。
一些体外研究表明bsTCE的活性与靶标表达水平相关,如针对癌胚抗原(CEA),CD33和人类表皮生长因子受体2(HER2;也称为ERBB2)的bsTCE所示。例如,与靶向产生促红细胞生成素的肝癌受体酪氨酸激酶A2(EPHA2)或前列腺特异性膜抗原(PSMA)的bsTCEs的这种相关性。尽管有这些相反的结果,但bsTCEs的细胞毒性活性可能需要一定的靶标表达阈值。每种抗原的这种阈值可能不同。例如,已经提出,靶向骨髓瘤抗原Fc受体样蛋白5(FcRL5)的少至50个bsTCE分子的结合足以通过FcRL5指导的bsTCE RG6160诱导有效的T细胞活化和靶细胞凋亡,而CEA bsTCE cibisatamab(Roche)的细胞毒活性至少需要10,000个CEA结合位点。
bsTCE的活性还取决于抗原的性质,而不是其在细胞表面的表达水平,例如其在膜中的迁移性。另外,已知TAA分子的大小和到靶细胞膜的表位距离严重影响bsTCEs的细胞毒性潜能。实际上,小的靶标大小和与近膜表位的结合使bsTCE能够建立紧密的细胞间膜-膜附近,从而导致最佳的突触形成和有效的T细胞介导的细胞毒性。
已经提出,由于结合亲和力的增加,使用二价靶向肿瘤的臂可以诱导增强的bsTCEs的效力和肿瘤选择性。正在开发几种四价bsTCE,包括上述的Adaptir和TandAb化合物,它们具有两个CD3和两个TAA结合单元。或者,正在开发对CD3单价和对TAA结合二价的三价1 + 2 bsTCE。例如,RG6026(也称为RO7082859; Roche)具有这种1 + 2设计,与其他bsTCE格式相比,可以提供更高的肿瘤抗原亲和力,T细胞活化和肿瘤细胞杀伤力。它由源自奥比妥珠单抗的两个抗B淋巴细胞抗原CD20 Fab,一个通过短的柔性接头与一个抗CD20 Fab融合的抗CD3 Fab和一个经工程改造以防止与Fc结合的异二聚体Fc域组成。受体和C1q。除了RG6026外,其他四个靶向CD20和CD3的bsTCE也正在I期临床试验中(GEN3013,mosunetuzumab,REGN1979和plamotamab; 表1),它们都是不对称的全长IgG双特异性抗体,设计为1 + 1。它们是使用不同的技术开发和生产的。值得注意的是,GEN3013的皮下制剂旨在降低峰值细胞因子水平,同时保持B细胞耗竭的功效,在这方面,它与其他CD20 bsTCE在开发中有所不同。看到抗体设计和配方的这些差异如何在临床中进行比较将是令人兴奋的。
使用bsTCE靶向血液学与实体恶性肿瘤
大多数临床阶段的bsTCEs正在开发中,用于治疗血液系统恶性肿瘤(67%; n = 29/43)(图3; 表1)。 尽管有些人提出了新的靶标,例如C型凝集素结构域家族12成员A(CLEC12A)(tepoditamab,MCLA-117; Merus),FcRL5(RG6160; Genentech / Roche)或G蛋白偶联受体家族C组5成员D( GPRC5D)(JNJ-64407564; Janssen),其中许多产品针对的是经过验证的众所周知的B细胞或髓样抗原,包括CD19,CD20,CD33,CD38(也称为ADPRC1),CD123或B细胞成熟抗原(BCMA; 也称为TNFRSF17)。
靶向同一靶标以治疗血液肿瘤的化合物之间存在惊人的重叠。除了上面已经讨论过的针对CD20的五种bsTCE之外,还有六种针对BCMA,五种针对CD123,四种针对CD33,二种针对CD38(表1)。尽管目标是相同的,但这些bsTCE产品之间的格式通常有所不同。例如,使用各种不同的格式来设计临床分期CD123 bsTCE,分别代表基于片段的格式(flotetuzumab,Macrogenics / Servier),四价对称格式(APVO436,Aptevo Therapeutics)和三种非对称格式(vibecotamab,Xencor / Novartis; Jansj Pharmaceuticals的JNJ-63709178;以及赛诺菲的SAR440234)。 JNJ- 63709178和vibecotamab由于可能与3级不良事件和试验中的死亡有关,因此已被FDA批准。对临床数据的进一步分析将揭示针对相同抗原的这些不同形式在安全性和功效方面的表现。
为治疗血液系统恶性肿瘤而开发的bsTCE的靶标也通常在正常的B细胞和浆细胞上表达,但是可以耐受这些细胞的消耗而不会引起严重的不良事件。相反,许多实体肿瘤抗原在关键组织中低水平表达,因此可能通过针对表达抗原的组织的靶向T细胞反应性诱导不良事件,从而使针对实体瘤的bsTCE的开发复杂化。此外,由于这些癌症特有的因素,针对实体瘤的抗体疗法的开发可能比针对血液恶性肿瘤的挑战更具挑战性,这些因素尤其包括免疫抑制性肿瘤微环境,肿瘤脉管系统紊乱以及抗体的肿瘤渗透受限(和效应细胞)。尽管如此,临床上仍在评估约14种bsT-CEs,这些bsT-CEs靶向HER2,表皮生长因子受体(EGFR)变体(v)III,PSMA和EpCAM等实体瘤抗原(表1)。
由于普遍缺乏与(啮齿类)抗原和有效T细胞的交叉反应性,因此在动物模型中同时解决功效和安全性的临床前研究非常复杂,但是在人源化中已观察到使用多种bsTCE在实体肿瘤模型中有希望的抗肿瘤活性鼠标模型。例如,靶向CEA的bsTCE甚至在浸润较差的肿瘤中也诱导了高度发炎的肿瘤微环境,并且在人源化小鼠(即异种移植有人结肠癌细胞系和人外周血的NOG小鼠)中表达CEA的肿瘤消退单核细胞(PBMC))。在已建立的皮下皮实体瘤模型中,针对人钙粘蛋白的bsTCEs还诱导了肿瘤特异性炎症,该模型中补充了经人腹膜内注射的人PBMC和Glypican 3(GPC3),并在人CD3转基因小鼠中注射了经人GPC3转染的鼠肿瘤细胞。有趣的是,在同系小鼠黑色素瘤模型中,以bsTCE为靶点的小鼠酪氨酸酶相关蛋白1(TRP1)和CD3小鼠治疗不会诱导细胞因子的释放和毒性,前提是bsTCE包含完全不含Fc-的惰性介导的效应子功能Fc片段。此外,bsTCE将已建立的黑色素瘤转变为炎症部位,不仅有T细胞流入,而且还有自然杀伤(NK)细胞和炎症巨噬细胞大量涌入。但是,bsTCE无法在该模型中诱导长期免疫,如未经治疗和长期存活的小鼠中相似的肿瘤生长率所表明的那样,该小鼠最初通过bsTCE治疗清除了肿瘤。
对bsTCE治疗的耐药性
bsTCE的抗肿瘤活性通常受到耐药性发展的限制。对潜在耐药机制的研究表明,肿瘤细胞上bsTCE特异性TAA的下调是肿瘤逃逸的机制之一。例如,在用blinatumomab治疗的ALL患者中注意到CD19复发,并阻止了bsTCE的进一步活性。
其他抗性机制可能涉及调节性T细胞或免疫检查点对免疫的抑制。例如,增强的程序性细胞死亡1(PD-1)和程序性细胞死亡1配体1(PD-L1)的表达可能受bsTCE治疗诱导,但限制了它们的活性,同时抑制了PD-1-PD-L1轴研究表明可增强bsTCE的临床前抗肿瘤活性。初步临床结果表明,CEA bsTCE cibisatamab与PD-L1抗体atezolizumab联合用于转移性结直肠癌,以及blinatumomab与PD-1抗体nivolumab联合使用的临床活性增强的早期迹象和可控的安全性在r / r ALL中。早期数据还表明,PD-1抗体西米普利单抗和CD20 bsTCE REGN1979组合使用的安全性可接受。但是,这些数据仍为时过早,后续的随访将揭示这种联合治疗策略是否可以改善患者预后。
激活T细胞的其他双特异性方法 Alternative bispecific approaches for activating T cells
抵抗免疫检查点的抗体的临床成功促使细胞毒性T淋巴细胞抗原4(CTLA4),PD-1和PD-L1,针对这些和其他免疫检查点的bsAb出现。反映出人们对这些免疫检查点抑制剂的浓厚兴趣,PD-1–PD-L1轴经常被靶向,至少有九种bsAb与CTLA4,淋巴细胞激活基因3(LAG3)或T细胞免疫球蛋白一起靶向其中一种抗原粘蛋白3(TIM3;也称为HAVCR2)现在处于早期临床研究中(TABle 2)。同时针对两个免疫检查点的理论依据是,结合单克隆抗体靶向这些检查点的联合研究中观察到的改善的临床获益。例如,与单独使用ipilimumab的治疗相比,使用ipilimumab(抗CTLA4)和nivolumab(抗PD-1)治疗黑色素瘤患者可提高生存率。然而,增加的抗肿瘤活性与明显增加的免疫相关不良事件有关。
为了提高PD-1和CTLA4联合靶向的安全性,已设计了Fc沉默的bsAb通过高亲和力的PD-1结合抑制PD-1途径,同时用低亲和力的结合臂抑制CTLA4。这种设计有利于在PD-1–CTLA4双阳性肿瘤浸润淋巴细胞中抑制CTLA4,同时减少与表达CTLA4的外周血T细胞的结合,这可能转化为更有利的安全性和耐受性。目前,在早期临床试验中正在评估四种PD-1×CTLA4 bsAb的安全性和早期疗效(表2)。阻断两种免疫检查点抑制剂的概念也在临床上针对其他靶标组合进行评估,例如PD-1×LAG3,PD-1×TIM3和PD-L1×CTLA4(表2),而许多其他药物正在临床前开发中。

从临床研究中可获得的数据将指导进一步优化针对免疫检查点的bsAb概念,同时确保可接受的安全性。预期靶向两种检查点阻断分子的bsAb的作用机理主要是组合的,因为通过结合两种mAb也可以轻松实现阻断活性。尽管如此,通过将低亲和力和高亲和力结合臂结合在单个分子中,例如,这些bsAbs可以被设计成具有专一的特征,目的是增加安全性或改善药代动力学。
或者,积极调控T细胞活化的途径,例如诱导性T细胞共刺激物(ICOS;也称为CD278)和肿瘤坏死因子受体超家族成员4(TNFRSF4;也称为OX40或CD134)途径,由bsAbs产生,包括一些临床上正在发展中(表1)。可以通过使用专心的bsAb概念来实现通过检查点激动剂对免疫细胞进行必要的条件激活。 INBRX-105(PD-L1×4-1BB;也称为TNFRSF9或CD137)是一个示例,旨在仅在遇到PD-的肿瘤环境中通过检查点激动剂分子4-1BB(重新)激活T细胞。 L1同时通过PD-1–PD-L1轴取消抑制。其他概念将免疫调节结合臂与靶向肿瘤的臂(如间皮素(MSLN))结合在一起。
超越经典T细胞:桥接其他效应细胞类型
尽管有其前景,基于CD3的bsTCE仍显示出许多缺点,包括潜在的高毒性,特别是对于具有广泛组织表达的靶标,例如EGFR和EpCAM。例如,Lutterbuese及其同事报道了食蟹猴中EGFR bsTCE的剂量反应非常陡峭,在高剂量时具有极高的毒性。进一步证明,EGFR bsTCE具有诱导EGFR旁观者细胞杀伤的能力。 Kebenko及其同事在I期临床研究中对实体瘤患者的solitomab(也称为AMG)(EpCAM bsTCE)的安全性和抗肿瘤活性进行了评估,并观察到了严重的不良事件,在所有剂量水平上均存在剂量限制的毒性,从而阻止了逐步升级达到治疗水平。基于CD3的bsAbs的一个特征是它们可以激活所有T细胞,而与谱系无关,这不仅会引起毒性,而且还会限制功效。例如,Duell及其同事证明了blinatumomab还可以激活调节性T细胞,从而抑制细胞毒性T细胞增殖和杀死肿瘤细胞。实际上,高调节性T细胞数量已显示出可预测r / r B细胞ALL患者对blinatumomab无反应。
这些观察结果支持以下观点:募集特定的T细胞亚群用于杀死肿瘤细胞可能比现有方法更具优势。 在这方面,Vγ9Vδ2T细胞特别受关注,因为它们代表了参与自然免疫监视的强大的促炎细胞。 单态的Vγ9Vδ2TCR感测到存在于肿瘤细胞中的,源自传染原或代谢异常的磷酸抗原。 Vγ9Vδ2T细胞普遍存在于大量血液和实体瘤中,并且它们在肿瘤中的存在与良好的预后相关。 已经报道了靶向HER2和EGFR的第一个双特异性γδTCE。 后者分子诱导了患者来源的结直肠癌细胞的裂解,并显示出对原发性EGFR +角质形成细胞的活性极低,因此有望增加治疗窗口
其他方法也侧重于重新定向和激活NK细胞,例如通过由针对CD16A(也称为FcγRIIIa)的单链可变片段(scFv)组成的三特异性分子,将其通过IL-连接到抗TAA scFv的NK细胞上15个链接器。此外,还显示了CD16×HER2双特异性分子通过表达CD16受体的NK细胞和γδT细胞诱导杀伤作用。 AFM13是针对CD16A和CD30的TandAb,它能够触发NK细胞介导的CD30 + NHL细胞杀伤。该bsAb由通过接头偶联的四个抗体可变片段的线性阵列组成,两个CD30结合位点位于CD16A的两个结合位点之间。肿瘤细胞CD30抗原因此与分子的中间部分相互作用,而分子的两端仍然可用于效应细胞的结合。应当指出,CD16A还在循环巨噬细胞和组织巨噬细胞上显示表达,因此作用机理不太可能是NK细胞特异性的。
最后,对CD47(也称为IAP)在调节效应细胞介导的杀伤作用中的作用进行了研究。 CD47通过抑制表达信号调节蛋白-α(SIRPα;也称为SHPS1)的效应细胞,作为吞噬作用的负调节剂。然而,CD47的普遍表达使其成为一个困难的目标。 Dheilly及其同事使用他们的κλbsAb平台开发了一种解决方案,其中将低亲和力的CD47抗体与高亲和力的抗肿瘤抗原抗体结合使用,从而确保CD47仅被bsAb结合在肿瘤细胞上-表达两种抗原。 CD47×CD19 bsAb可诱导概念性验证,可诱导Fc介导的吞噬作用增强,并在存在大量与肿瘤无关的CD47时保持其活性。有趣的是,由于仅通过组合亲本抗体无法实现相似的肿瘤特异性,亲和力工程化方法可生成靶向同一细胞上两个分子的专性bsAb。
超越肿瘤学:传染病中的T细胞重定向
病毒特异的bsTCE设计用于将CD8 + T细胞重定向至表达病毒表面抗原的感染细胞,并且已在乙型肝炎病毒,巨细胞病毒和HIV-1感染中进行了描述。对于HIV而言,gp120包膜糖蛋白(Env)特异的bsTCE(主要以基于片段的1 +1形式评估)能够在体外诱导被多种HIV分离物感染的细胞(CD4 + T细胞和巨噬细胞)的杀伤作用。并且显示在连续抗逆转录病毒治疗中抑制了从受试者体内获得的HIV感染细胞中的HIV复制。另外,潜伏感染细胞上存在的CD3受体的结合可以重新激活病毒复制,从而诱导病毒抗原的表达,并使CD8 + T细胞也能够杀死这些细胞。这些研究共同表明,HIV特异的bsTCE具有治疗性体内清除HIV的治疗潜力,特别是与补充干预措施(如潜伏期逆转剂以解决病毒库)相结合时。在I期研究中,一种针对bsTCE的MGD014(非对称1 +1格式)正在接受HIV感染的个体进行抗逆转录病毒治疗(表3)。

肿瘤以外:再生医学中的T细胞重定向
在缺血性再灌注(IR)损伤后静脉内施用干细胞和祖细胞以恢复组织损伤,已在急性心肌梗死模型中显示出治疗潜力。然而,这些细胞对损伤部位的低归巢效率被认为是成功进行临床翻译的主要限制之一。人们认为增强干细胞和祖细胞的靶向递送可以提高组织再生的效率,因此目前正在研究基于bsAb的重定向方法。
IR损伤后,由于活化的血小板在心脏中积聚,因此将这些血小板靶向输送具有再生潜能的细胞。在这种情况下,已经研究了主要血小板整联蛋白糖蛋白(GP)GPIIb / IIIa(也称为αIIb/βIIIa或CD41 / CD61)或CD41的活性构型作为靶向抗原。 bsAb介导的内皮祖细胞通过与干细胞标志物CD34结合并与CD41 +血小板结合,在急性心肌梗塞的小鼠模型中实现了有效的心脏修复,这一点已通过心脏功能,心脏形态和免疫组织化学方法得到了证实。通过靶向干细胞抗原1的bsAb(SCA1,也称为LY6A.2 / LY6E.1;鼠类造血干细胞标志物)和激活的GPIIb /构象,将PBMC的一部分传递至IR损伤部位。血小板上的IIIa导致浸润性炎性细胞显着减少。这些SCA1 + PBMC可以通过调节心脏修复机制来减少纤维化,增加毛细血管密度和恢复心脏功能。
2.2 桥接受体(顺式结合) Bridging receptors (in-cis binding)
对EGFR和HER2等致癌受体酪氨酸激酶(RTK)的靶向抑制是一种成功的抗癌方法,但耐药性的发展是此类疗法的主要局限性。耐药性通常涉及其他RTK的上调,这些RTK绕过特异性受体抑制作用以激活平行的信号通路。例如,酪氨酸蛋白激酶MET通路的上调或激活赋予非小细胞肺癌(NSCLC)肿瘤以EGFR酪氨酸激酶抑制剂治疗的抗性。这为共同靶向多种RTK的bsAb的开发提供了理论基础(图5b),其中许多正在临床研究中(表1,2)。尽管靶向多个RTK是一个组合概念,但是某些分子显示出专一的特征。例如,JNJ-61186372(Janssen Pharmaceuticals)是通过cFAE生成的EGFR×MET bsAb,可通过抑制配体诱导的激活和受体降解来阻断EGFR和MET信号传导。 JNJ-61186372具有抗体依赖性细胞毒性(ADCC)活性,通过产生具有低岩藻糖Fc碳水化合物的抗体可以提高该活性。除了ADCC,Fc相互作用似乎是EGFR和MET下调所必需的。最近,据报道对EGFR驱动的NSCLC具有初步的临床活性,而安全性可控。
无法根据亲本抗体的特性预测最有效和最有效的双特异性抗体。因此,通常通过功能筛选由抗体对基质产生的bsAb的面板来经验选择最佳的bsAb。 JNJ-61186372基于优异的功能活性,从一组EGFR×MET bsAbs中选择。选择最佳Fab臂组合的重要性通过以下事实证明:JNJ-61186372是筛选中唯一具有所有所需活性,包括不存在不希望的MET和/或EGFR受体活化的bsAb。 JNJ-61186372确实代表了一个有趣的例子,说明bsAb的活性可能不同于亲本抗体的混合物。二价MET抗体诱导MET交联和肿瘤细胞活化,从而阻碍了它们作为癌症治疗剂的用途。非对称1 + 1设计中的单个(非激活性)MET结合臂与合适的EGFR结合臂的结合,确保了能够阻断EGFR和MET信号传导的分子的产生。在另一个实例中,对545份针对HER2和HER3的bsAb进行了无偏见的表型筛选,鉴定出在体外和体内均能有效抑制肿瘤细胞生长的HER2×HER3 bsAb(PB4188)。该抗体以高亲和力与HER2结合,从而增加了HER3 Fab的局部浓度,即使在高HRG浓度下,也导致与HER3的结合以及调蛋白(HRG)介导的HER2和/或HER3信号传导的抑制。相比之下,HER2和HER3 mAb的组合在高HRG浓度下无法抑制HER3信号传导。 Zenocutuzumab(MCLA-128)是源自PB4188的ADCC增强型临床候选药物,目前正针对各种已报道HRG-HER3途径活化的适应症进行临床评估(表1)。
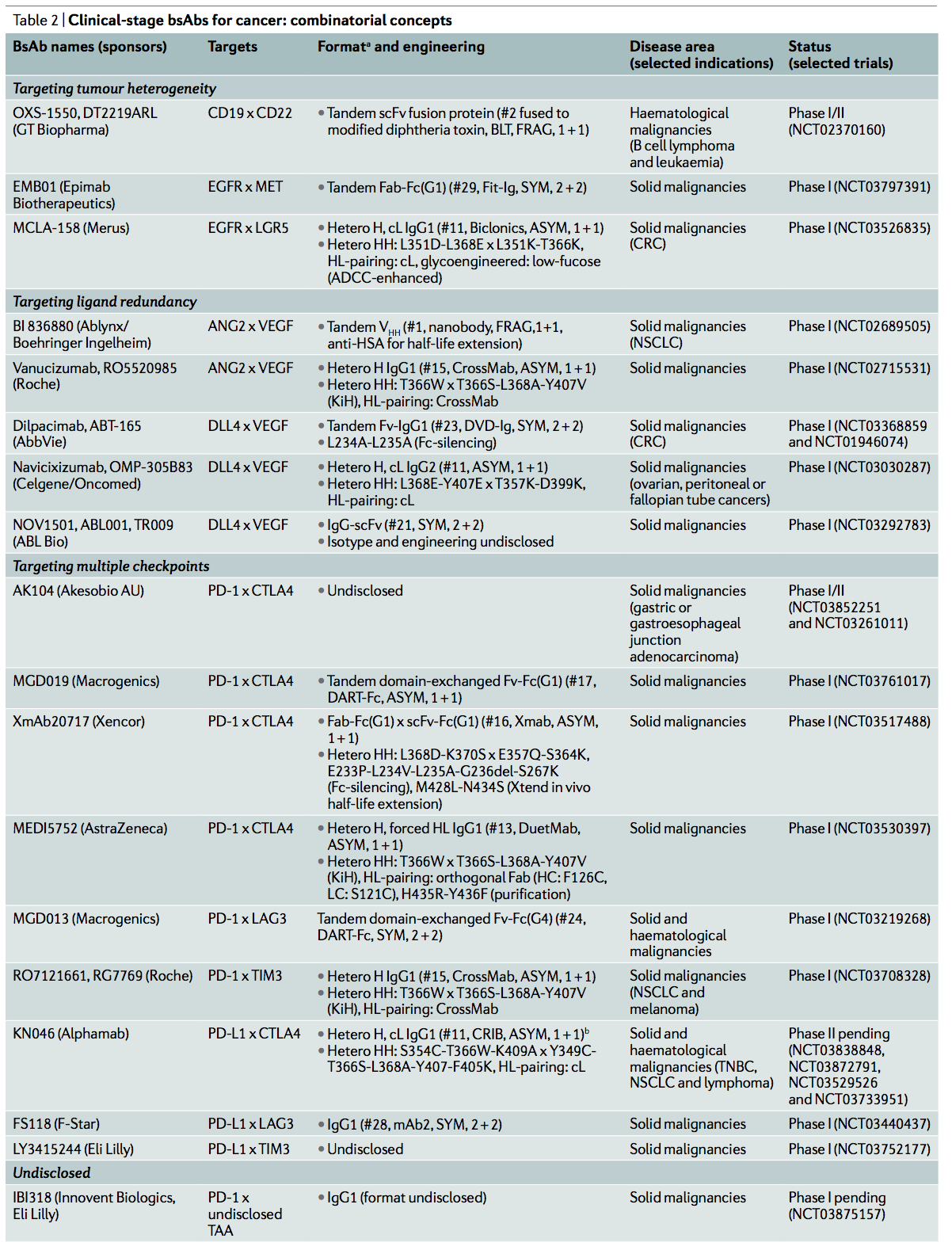
2.3 靶向配体冗余 Targeting ligand redundancy
针对多种生长或血管生成因子的靶向冗余代表了bsAb的关注领域。该组中的bsAb本质上是组合的。 Dilpacimab(也称为ABT-165)是一种具有双重可变域(DVD-Ig)的bsAb,该双域结合血管内皮生长因子(VEGF)和δ样配体4(DLL4),其发展最快。该药物处于一项II期临床试验中,正在与之前接受过治疗的转移性结直肠癌患者同时接受化疗的患者的抗VEGF mAb贝伐单抗进行比较(表2)。
在非癌症适应症中,正在对患有弥漫性皮肤系统性硬化症的患者进行的II期研究(表4)中评估了由抗IL-4抗体与靶向IL-13的可变域融合而成的romilkimab。在III期研究中,唯一的bsAb是Faricimab(也称为RO6867461),是一种基于人IgG1的CrossMab,它的一只臂结合VEGF,另一只臂结合血管生成素2。构成该分子的所有四个蛋白质链都是不同的。另外,为了优化用于眼科适应症,对Fc区进行修饰以消除与包括FcRn的IgG Fc受体的结合,从而消除了效应子功能并增加了全身清除率。 Faricimab目前正在进行糖尿病黄斑水肿患者的两项III期研究,先前已在新血管性年龄相关性黄斑变性的3期II期研究中进行过评估(表4)。faricimab治疗的糖尿病性黄斑水肿视力丧失的患者与基线相比具有临床意义的视敏度提高,与经批准的抗VEGF mAb片段ranibizumab治疗的患者相比,视敏度具有统计学意义。
2.4 双参数bsAb Biparatopic bsAbs
可以将bsAb设计为同时结合同一靶标上的两个不重叠的表位,而不是靶向两种不同的蛋白。双原位靶向可通过抗原交联和聚集增强结合强度,从而模仿抗体混合物和多克隆抗体的效果。因此,双参数bsAbs本质上是组合概念。
ZW25是一种bsAb,旨在与HER2双位结合,以增加亲和力,从而使HER2信号转导更有效地沉默(表2)。此外,它还可以从细胞表面去除HER2蛋白,并增强了效应子功能。这些综合的作用机制在临床前模型中转化为抗肿瘤活性,其作用比曲妥珠单抗140诱导的更有效。初步临床数据显示,对于患有晚期HER2 +癌症(包括胃食管癌和乳腺癌)的患者,其安全性可控且有望获得早期疗效。如Li和同事所述,基于ZW25骨架的双对位抗体-药物偶联物也正在开发中,它可能会通过改善溶酶体的内在化和降解而得益于更有效的毒素释放。
2.5 激动性双特异性抗体 Agonistic bispecific antibodies
与通过抑制性抗体阻断致病性信号传导相反,一些治疗概念要求通过激动性抗体激活受体信号传导,例如以上针对检查点激动剂所描述的。专性bsAbs也特别适合激活多组分受体复合物,其中激活需要结合受体和共受体(图5c)。
据报道,成纤维细胞生长因子21(FGF21)途径的激活可改善肥胖症和糖尿病。然而,重组FGF21具有不良的药代动力学性质,并且长期治疗具有副作用的风险。因此,先前设计的bsAb BFKB8488A(Roche)通过选择性靶向成纤维细胞生长因子受体1C(FGFR1C)-β-klotho(KLB)受体复合物来激活该代谢途径。尽管KLB在肝,脂肪和胰腺组织中选择性表达,但FGFR1C具有广泛的组织分布。因此,这些受体的共同靶向可能仅将信号激活限制于共同表达KLB和FGFR1C的组织,并限制广泛FGFR激活的不良后果,例如诱导细胞增殖。一项正在进行的首次人体试验的初步结果显示,患有胰岛素抵抗的肥胖受试者的心脏代谢特征得到改善(表3)。
2.6 辅因子模拟物
针对充当酶或辅因子的酶-底物复合物的BsAbs提供了强大而具有挑战性的明确机会,在此机会中,空间方面(即酶和底物的最佳位置)至关重要(图5d)。 Sampei及其同事着手产生一种bsAb替代FVIII作为A型血友病的潜在治疗方法,以防止FVIII功能障碍引起的出血发作。设想bsAb模仿FVIIIa(FVIII的活化形式),具有将FIXa和FX结合在一起并随后增强FIXa催化活性的能力。经过广泛的筛选工作,从针对FIXa或FX的200 mAb中产生了40,000个不对称bsAb,导致具有FVIII模拟活性的bsAb命中率约为0.3%。为了解决链关联问题,每个命中仅用一条同源的L链表达,然后选择最有效的bsAb,并通过构架区和互补决定区(CDR)改组进一步优化常见的L链。其他几轮优化包括人源化以降低免疫原性风险,CDR诱变以进一步提高酶活性,可变区的电荷工程以改善溶解度和药代动力学以及等电点工程以促进bsAb从离子交换纯化中从同二聚体污染物中分离出来。最终的候选药物称为ACE910或艾米珠单抗,对FIXa和FX和/或FXa均表现出微摩尔结合亲和力,因此在血浆中只有一小部分在药物的有效剂量下形成活性三分子复合物(每毫升50-90μg) 。事实证明,使用艾米单抗预防A型血友病可有效减少有或无抑制剂(即抗FVIII抗体)患者的出血。 Emicizumab在2017年获得FDA的常规预防,以减少具有FVIII抑制剂的A型血友病患者的出血(表3)。 2018年10月的进一步批准还包括在没有FVIII抑制剂的患者中,使用Emicizumab预防。 Emicizumab于2018年3月在欧洲首次获批。
2.7 搭载方式 Piggyback approaches
仅将bsAb的第一特异性用作第二特异性的运输方式的方法本质上是专心的(obligate),需要顺序结合。这些方法被称为“背负式”(piggyback)或“劫持式”(hijacking)方法,旨在获得进入(或逃脱)受限(蜂窝)隔室的权限。 Raso及其同事描述了使用劫持概念进行蛋白质特异性递送的第一个例子,他们证明了通过介导毒素的内在化作用而对B细胞受体和蓖麻毒素A进行化学连接的双特异性Fabs诱导了毒性。后来的研究表明,具有bsAbs的(突变)白喉毒素的传递具有pH依赖性的结合,可用于增加内体中毒素的释放(在低pH下),从而改善细胞杀伤力。
最近的一个例子是劫持了转铁蛋白的胞吞途径,以穿越血脑屏障并进入免疫弱势的脑室(图5e)。通过用bsAb的一个结合臂靶向转铁蛋白受体(TfR),研究人员已证明,在阿尔茨海默病的临床前小鼠和猴模型中,β-分泌酶1(BACE1)特异的第二个结合臂的大脑递质得到增强。 。使用TfR亲和性优化的不对称1 +1格式,TfR×BACE1 bsAb可降低脑组织和脑脊髓液中BACE1的酶促产物淀粉样β(Aβ)肽水平。同样,在阿尔茨海默病病理模型的慢性小鼠中,不对称的TfR×AβbsAb(设计为1 + 2)可显着减少大脑皮层和海马中的斑块数量。
在埃博拉病毒(EBOV)感染中已经描述了第三种搭载方法,其中存在于未裂解的EBOV GP上的一个广泛保守且暴露于细胞外的表位,旨在在病毒摄取期间进入内体区室。隔离在晚期的内体中,GP的蛋白水解裂解揭示了进入细胞质所需的尼曼-皮克C1(NPC1)细胞内受体的隐性受体结合位点(RBS)。因此,通过GP特异的第一个结合域通过内体递送针对隐性RBS或NPC1的广泛中和的第二个结合域,Wec及其同事证明了bsAbs可以在体外中和所有已知的EBOV菌株,并且可以赋予暴露后预防小鼠中多种病毒的致命攻击。对称2 + 2和非对称1 +1格式都被证明是有效的,尽管1 +1设计显示出降低的效能,这可能是由于亲和力的损失。
Psl×PcrV bsAb MEDI3902是一种全长IgG1抗体,具有以对称2 + 2形式插入Fab和Fc之间的scFvs(表3),已显示出采用类似的背负机制来增强对铜绿假单胞菌的杀灭作用。由嗜中性粒细胞引起(图5f)。该bsAb靶向铜绿假单胞菌血清型独立的III型分泌系统毒性因子的持久因子Psl和针尖蛋白PcrV。 Psl是一种主要的细胞外多糖和铜绿假单胞菌生物膜成分,与抑制补体沉积,减少嗜中性粒细胞对细菌的识别和吞噬有关。 MEDI3902对Psl的抑制确实导致嗜中性粒细胞的内在化增加。 PcrV在减少吞噬体酸化和杀死细菌方面发挥作用。 PcrV由内化细菌表达,是III型分泌物注射体的一种成分,在其中它通过防止转移到酸性液泡而在铜绿假单胞菌的存活中起作用。 MEDI3902的抗PcrV臂抑制了PcrV活性,并增加了所摄入细菌对此类低pH囊泡的定位。因此,bsAb中抗Psl和抗PcrV之间的物理连接通过经由抗Psl结合臂piggy带在细菌上,有助于增加抗PcrV的获取。在细菌感染的小鼠模型中,与抗PcrV和抗Psl抗体的组合相比,bsAb提供了增强的保护作用,表明双特异性确实具有机制上的优势。 MEDI3902还提供了针对缺乏PcrV毒力因子的菌株的保护作用的观察结果表明,这种专一性并非绝对的。
通过参与溶酶体运输的双重靶向CD63(也称为LAMP3)和肿瘤特异性的模型抗原HER2,可以改善抗体-药物偶联物向溶酶体的细胞内递送。在这种情况下,使用肿瘤特异性臂(HER2)和低亲和力的CD63结合臂来确保肿瘤特异性(另请参阅交叉臂结合效率)并降低毒性。确实,通过以这种方式劫持溶酶体的运输,研究人员证明了在异种移植小鼠模型中,无论是体内还是体外,只要两个靶都被表达和靶向,功效就可以证明。
最后,通过双重靶向或多重靶向逃避内体区室代表一种背负式方法,该方法适用于基于片段的形式以克服Fc区的缺乏。例如,vobarilizumab和ozoralizumab是基于H链的仅可变域(VHH)的bsAb,目前处于II期研究中,分别靶向IL-6受体和肿瘤坏死因子(表3)。为了延长半衰期,这些分子还靶向HSA,以劫持FcRn挽救途径并逃避溶酶体降解,这是抗体Fc区通常提供的特性。专性指定可以说是任意的,因为这些bsAb的持续功能性也可以通过连续给药(如blinatumomab)或通过用Fc片段表达串联VHH来获得。
三、 推进双重目标概念 Advancing dual-targeting concepts
与治疗性单克隆抗体相比,与双重靶向概念相关的增加的复杂性在发现和开发的不同阶段可能会带来其他挑战。 本节将讨论在解决某些挑战方面的最新进展,这将有利于bsAb的整体发展。
3.1 格式和设计如何影响开发策略 How format and design affect development strategy
bsAb格式类别可以影响在双重靶向概念中探索的抗原结合域的功能,反之亦然。例如,需要共同的L(cL)链或cH链的形式可能会将(预先存在的)抗体组的使用限制为那些可以容忍这些限制的组合。因此,已经开发了互补技术,例如cL链转基因动物和cL链或cH链噬菌体展示文库,以弥补这些局限性并在发现阶段增加抗体库。同样,抗体片段在重新格式化后可能无法始终如一地产生具有相似(功能)特性的全长IgG,从而限制了它们在大多数不对称形式中作为抗体来源的价值。但是,抗体片段与文库选择方法的相容性可用于定制的抗体特性(例如亲和力成熟,调节物种交叉反应性和消除潜在的制造责任)在开发过程中代表了一个非常吸引人的特征。为了利用基于全长抗体的格式进行文库选择的优势,正在探索在发现流程的早期整合整个抗体片段库的重新格式化的策略。
需要同时绑定到两个目标的双重目标概念的开发并不简单,因为设计参数(例如亲和力,价,表位特异性和格式体系结构)可能显示出相互依赖性。例如,当在同一细胞上靶向两种膜抗原时,靶标结合的价位不仅可以通过亲合力影响单特异性相互作用,而且可以通过跨臂结合效率影响与第二种特异性的相互作用。此外,格式结构和相对互补位的取向还可以影响最佳靶标结合(取决于相对表位拓扑或靶标分布),并可能影响治疗活性。因此,对于这些应用,结合臂的正确组合的选择(和优化)可能需要针对正确的生物学活性筛选多达数千对结合对。这需要一个集成的发现过程,该过程通过经验选择策略来询问设计参数和目标绑定库的全部数组,以帮助成功识别最有效的bsAb。以最终产品格式或易于适应最终产品格式的格式进行筛选被认为可以进一步加快成功开发的速度
相比之下,在两种特异性的治疗活性依次发生且不需要同时结合的应用中,相对的互补位取向可能起次要作用,因此可以通过不同设计的bsAb格式解决。而且,两种特异性的独立活性使得在发现阶段可以选择和优化单个结合臂,然后在不同的bsAb格式类别中进行设计重组,并根据所需的最终产物要求选择最终产物。
影响bsAb格式选择的其他考虑因素可能包括与其他IgG同种型骨架(例如IgG2或IgG4)一起使用以减少Fc介导的毒性的格式的灵活性。此外,bsAb格式与基于常规抗体治疗方法建立的其他优化策略的相容性,可进一步促进其成功开发。在这种情况下,可改变药物代谢动力学特性,调整功能特性(例如,糖工程以增强ADCC或引入突变以沉默效应子功能)的Fc工程策略或促进制造过程(例如等电点工程或去除)电荷异质性)。
3.2 转换工具以增加成功 Translational tools to increase success
在体内建立概念验证是治疗性(bs)Abs临床开发转化阶段的重要一步。但是,随着概念的日益复杂(双重靶向),完整的免疫系统和疾病组织微环境(包括与疾病相关的免疫细胞)的存在对于准确捕获要靶向的相关生物学至关重要。结果,经常在免疫能力强的小鼠中使用同系模型与(替代)小鼠抗体相结合来研究这些复杂概念的功效和安全性,以最大化(潜在)利用效应子功能并避免抗药物抗体反应。另外,正在探索基因工程化的小鼠模型,其中引入了相关的人类靶标或被其人类对应物替代的等效小鼠靶标,以补充体内验证工作。
对于替代性bsAb,基于片段的形式可轻松用于翻译研究中。然而,它们不良的药代动力学特性和缺乏Fc介导的效应子功能将其翻译价值限制在可耐受这些限制的治疗概念上。在单个鼠类亚类的基础上,替代对称和非对称bsAb格式有望具有规则的药代动力学特性,并可能具有最天然的功能特性。研究人员使用这种替代性bsAb对T细胞重定向的不同翻译方面进行了建模,例如对惰性骨架的要求,CD3亲和力对生物分布的影响,T细胞募集的机制或联合疗法的原理
3.3 超越双重目标
为双特异性分子增加额外的特异性可以引入额外的功能,从而有可能增加其治疗活性。 因此,正在不同的治疗领域探索包含三个或更多不同抗原结合位点的基于多特异性抗体的形式。
从设计的角度来看,创建多特异性的最直接方法是将其他抗体片段设计成现有的基于片段或对称的bsAb格式。 但是,也可以将对称设计与不对称H链异二聚或强制HL链配对策略结合使用,以引入额外的特异性。 还描述了更精细的工程设计,例如将多个强制HL链配对策略组合到一个分子中
在肿瘤学中,同时靶向更多介导疾病的受体或串扰性信号传导级联被认为增加了有效解决受体冗余或异质性的机会,并降低了逃生的风险。因此,许多多特异性形式都采用了这一概念,并针对同一受体家族的多个成员,例如EPHA2,EPHA4和EPHB4或EGFR和HER3与胰岛素样生长因子1受体(IGF1R)和MET或HER2和VEGF。但是,与bsAbs一样,人们可能会争辩说,这些概念并不是严格必须的,可以通过联合疗法来实现。但是,从发展的角度来看,一个分子的法规批准比寻求每种抗体的批准更快(且更具成本效益)的论点与多特异性形式更相关,但仍应弥补缺乏特异性的不足。自由分配单个成分以获得最佳功效。例如,在临床上通过西妥昔单抗和贝伐单抗的联合治疗观察到毒性显着增加,而功效有限,这突出了在多特异性形式下可能难以解决的给药挑战。
还正在探索多特异性作为病毒性疾病的治疗选择,以覆盖遗传多样性并预防获得性耐药。 在HIV-1感染中,与单一的广泛中和抗体或bsAbs的组合相比,三特异性(1 +1 + 1)形式具有更高的中和效力和广泛的覆盖范围(> 98%覆盖率)。 在EBOV感染中获得了相似的结果。 一项HIV-1研究进一步证明了使用三特异性抗HIV-1抗体对非人类灵长类动物中两种差异敏感的嵌合猿猴-人(SH)IV分离株混合物的体内保护作用。
3.4 即将发生什么? What is on the horizon?
展望未来,目前处于发展初期的有前途的概念创新代表了不久的将来令人振奋的可能性。 由Keyt和同事率先提出的这种新颖概念的一个例子,是利用带有J链的抗体类别(例如IgM和IgA)的天然结构构建的,该链上附着了效应细胞靶向臂。 对于五聚体IgM,这将允许双特异性格式具有1 + 10设计的格式(对于二聚体IgA,则具有1 + 4设计格式的格式)可以介导以极低水平表达的病原性驱动程序的高亲和力靶向。 但是,工业规模生产IgM仍然在技术上具有挑战性。
另一个有前途的概念是使用mRNA编码或DNA编码格式的治疗性bsAb的非蛋白质递送。例如,Stadler及其同事在体内持续生产了优化的,经核苷修饰的mRNA,其编码基于片段的bsTCE(针对CD3和紧密连接蛋白claudin 6(CLDN6)的1 +1设计)。通过基于聚合物和/或基于脂质的制剂,研究人员确保了静脉内给药后在肝脏中的靶向和翻译,并证明与相应的纯化蛋白递送的bsAb一样有效地消除了晚期肿瘤。同样,Petal等人在肌肉内给药和电穿孔后显示出DNA编码的对称bsAb格式的体内持续表达。在这项研究中,MEDI3902的DNA递送版本(针对铜绿假单胞菌蛋白PcrV和Psl的对称2 + 2设计;与上文讨论的蛋白质相比)显示出难以区分的效力,并在肺炎小鼠模型中具有免受致命攻击的保护作用。由于药物级mRNA和DNA的生产速度很快,研究人员认为这种方法可以加速新型bsAbs的临床开发。此外,在DNA的情况下,温度稳定性将使其能够长期保存,更容易运输,从而可以管理更广泛的人群。 DNA介导的抗体转移的概念在供应链的便利性,体内长期抗体生产的潜力和成本效益方面,对于传染病的应用特别有希望。基因转移的专一性bsAbs为靶向病毒或细菌的脆弱性提供了新的机会,因此可以为针对挑战性微生物的疫苗提供有用的替代品。
在另一种有趣的组合方法中,显示了将带有bsTCE转基因(靶向CD3和EGFR)的溶瘤腺病毒与叶酸受体-α(FRα)特异性的CAR T细胞组合在体内具有协同抗肿瘤作用。 在这项研究中,Wing及其同事证明了在癌症小鼠模型中增强的T细胞活化和生存期延长,为对该概念进行进一步的临床评估提供了依据。
四、结论
治疗性bsAb是一组迅速发展的多样化分子。 两种bsAb已获得监管部门批准,目前已上市:blinatumomab,一种基于片段的bsTCE在癌症中的应用; Emicizumab,一种具有自然结构的全尺寸双特异性IgG,可在出血性疾病中用作酶辅因子的替代品。 在各种适应症的临床研究中,超过85种bsAb正在进步。 当前,人们高度关注癌症,这可以部分归因于bsAbs在免疫肿瘤学方法中的巨大潜力,其中有43个bsTCE和15个bsAb在开发中靶向免疫检查点分子。
BsAb的形式多种多样(图2),它们会影响生产,化合价,Fc介导的效应子功能和体内半衰期。因此,正确的bsAb格式的选择强烈地取决于所需的目标产品概况和临床适应症。尽管种类繁多,但从机理角度对bsAb进行分类似乎非常有用。 BsAb设计可以是组合的,其中bsAb可以用作抗体混合物的替代品(例如,以降低成本),但是这种方法的缺点是抗体结合域的比例在早期固定。因此,毒性,药代动力学和药效学不能通过不协调的剂量来优化。专性bsAb是结合域的物理连接产生新功能的bsAb。即,抗体混合物无法实现的功能。在暂时性闭塞中,作用机制是由顺序结合事件介导的,其中第一结构域的结合促进了第二结构域的活动,例如,通过提供对远端位点的进入。在空间上,作用机理取决于同时结合,其中bsAb介导其定位,例如,将效应细胞靶向肿瘤或酶靶向其底物。空间专一性的设计特别具有挑战性,因为它需要两个结合域及其目标表位的精确3D定位。可能需要对大型bsAb文库进行无偏见的表型筛选,才能获得最佳候选物。然而,应该指出的是,bsAbs之间的机制区别不是绝对的,因此在发展中,bsAbs代表了从组合机制到时空专性机制的频谱。
总之,bsAb为新颖的药物设计和开发提供了令人兴奋的机会,并且我们期望上述平台和概念的持续发展将提供持久的治疗效果。
参考资料
- Labrijn, A. F., Janmaat, M. L., Reichert, J. M., & Parren, P. W. H. I. (2019). Bispecific antibodies: a mechanistic review of the pipeline. Nature Reviews Drug Discovery, 18(8), 585–608. https://doi.org/10.1038/s41573-019-0028-1
