【4.4.2】预测MHCII的process过程(MHCII-NP)
官网:http://tools.immuneepitope.org/mhciinp/
CD4 + T细胞在调节免疫反应中起主要作用。通过识别主要通过主要组织相容性复合物(MHC)II类途径,从外源抗原产生的肽来激活它们。表位的鉴定很重要,表位的计算预测被广泛用于节省时间和资源。尽管有算法可预测肽与MHC II分子的结合亲和力,但尚无准确的方法可预测由于天然抗原加工而产生的配体。我们利用质谱法鉴定了从表达MHC II类的细胞中洗脱的肽,鉴定了大约14,000个天然加工的配体的数据集,以研究可能与从其源抗原释放出呈现的肽的裂解机制有关的序列标记的存在。该分析揭示了围绕配体的N-和C-末端的优选氨基酸,表明序列特异性切割偏好。我们使用这些切割基序开发了一种预测天然加工的MHC II配体的方法,并验证了它具有从独立研究中鉴定配体的预测能力。我们进一步证实,基于裂解基序的配体预测可与MHC结合预测结合,并且该结合预测具有优越的性能。但是,当尝试单独或与MHC结合预测结合预测CD4 + T细胞表位时,基于切割基序的预测未显示预测能力。假定基于CD4 + T细胞反应性鉴定为表位的肽通常没有明确定义的末端,则可能存在基序,但位于映射的表位之外。我们在计算上考虑到这一点的尝试并未显示出在充分表征的CD4 + T细胞表位周围存在切割基序的迹象。尽管我们尝试将MHC II配体洗脱数据中的裂解基序转化为T细胞表位预测的尝试可能不是最佳的,但其他可能的解释是裂解信号过分稀释而无法检测到,或者洗脱数据中富含产生的配体通过抗原加工和呈递途径,这种途径很少用于T细胞表位。
一、前言
主要的组织相容性复合物(MHC)I类分子实际上在所有有核细胞中都有表达,它们的主要生物学功能是将来源于加工内源性抗原的肽呈递给杀伤性CD8 + T细胞(1)。相比之下,MHC II类分子主要由专业的抗原呈递细胞(APC)(例如巨噬细胞,树突状细胞和B细胞)表达, 并且主要参与通过抗原呈递的内吞途径将外源抗原产生的肽与CD4 + T细胞结合和呈递(2)。当由炎症信号诱导时,非专业APC如内皮细胞,成纤维细胞,上皮细胞和肿瘤细胞也表达MHC II分子(3)。在抗原呈递的主要内吞途径中,细胞外抗原通过吞噬作用,巨胞吞作用或受体介导的内吞作用而被内在化,并在酸性和蛋白水解区室(如溶酶体或晚期内体)中被通常称为组织蛋白酶的蛋白酶降解(4)。较少见的是,MHC II分子还结合通过自噬获得的内源性抗原(即胞质和核蛋白)加工产生的肽(5)。
由与MHC II分子结合的抗原加工产生的肽复合物被转运到细胞表面,在那里它们可被CD4 + T细胞识别。 MHC II类多肽的CD4 + T细胞识别肽在多种免疫反应中起着关键作用,例如针对病毒和细菌感染以及寄生虫感染以及与过敏反应有关的免疫。除了这些公认的作用外,还应理解,MHC II类抗原呈递途径还涉及自身免疫(1、6)和癌症免疫(7-9)。这种广泛的免疫功能使识别被识别为CD4 + T细胞表位的MHC II类限制性肽成为重要的研究领域。
对于I类MHC限制性T细胞表位,I类MHC结合预测有助于T细胞表位的鉴定,从而大大减少了必须测试的肽的数量, 因为绝大多数T细胞表位都结合了预期结合肽的前0.5–2%(10)。相比之下,对于II类MHC限制性T细胞表位,结合预测不那么可靠(11),并且表位通常在预测结合物中的前10%或20%。部分原因是与I类MHC相比,II类MHC的预测配体亲和力更不可靠。II类MHC的结合预测已变得越来越可靠(12-16),但具有预测II类MHC的能力T细胞表位仍然比I类MHC差得多。这表明,超出MHC II类结合亲和力的因素有助于使肽被CD4 + T细胞识别。
众所周知,天然加工决定了哪些肽可用于结合MHC分子并随后呈现给T细胞,并且结合MHC分子的能力是免疫原性的必要但非充分必要。有几种算法可以帮助预测哪些肽是天然的I类配体(17、18)。天然配体最近也已用于训练I类预测算法(10、19、20),从而导致配体和CD8 + T细胞表位的整体性能得到改善。相比之下,目前尚无算法可预测II类MHC抗原呈递途径天然加工了哪些肽。合理的假设是,对MHC II类抗原加工和所得天然配体的分析可导致改进表位预测算法,并为加工机理本身提供更多参考。
基于质谱(MS)的技术的最新进展已导致鉴定出从MHC分子洗脱的大量肽(21-25)。 这些天然加工的肽组,称为人白细胞抗原(HLA)配体组或HLA肽组,是扩展有关抗原加工机理的现有知识的宝贵资源。 在这项研究中,我们分析了由MS鉴定的天然加工的II类MHC配体集,这些配体由表达MHC II类的细胞洗脱并从免疫表位数据库(IEDB)下载的肽(26)进行研究,以研究II类MHC抗原的加工机理和 检查该信息是否可用于改善CD4 + T细胞表位的预测。
二、材料和方法
3.1 配体数据的收集和筛选
通过使用以下标准通过查询IEDB数据库(26)收集主要的组织相容性复杂的II类配体洗脱测定数据:“仅阳性测定,表位结构:线性序列,无T细胞测定,无B细胞测定,MHC配体测定:MHC配体洗脱试验,MHC限制类型:II类。”收集的数据包括配体序列,配体起源的来源的详细信息,包括来源蛋白质名称,来源蛋白质中的配体位置,来源生物名称以及配体的限制性等位基因。使用Python脚本解析导出的数据。配体的来源序列是从UniProt收集的
在研究中使用了两组独立的配体数据。截至2016年7月,收集了第一个数据集,其中包含IEDB报告的35,367个配体。基于诸如配体长度和等位基因分布的因素筛选收集的数据。原始数据集中的配体长度在3到46之间,并且配体的分布随长度变化很大(图1A)。最丰富的长度是15,其次是14和16。这三个长度合起来几乎构成了数据的50%。从最初的数据集中,首先我们选择长度至少占总配体0.5%的配体,其中长度为9-23。这占收集的总数据的98.22%。接下来,分析数据集中配体的限制性等位基因,并选择明确列出等位基因的条目。例如,排除了其中仅将HLA等位基因列为基因位点的某些条目(HLA-DQ和HLA-DR),或我们没有结合预测算法的鸡,马,牛和小鼠等位基因。这样做是为了简化分析。将代表性的等位基因分配给指示血清型的条目(例如,对于HLA-DR7为DRB1 * 07:01)。具有相同配体序列和等位基因的冗余条目被删除。从该组中,排除了少于50个条目的等位基因。某些未明确标明来源蛋白的条目也被排除在外。我们进行了进一步的分析,以确定最佳的配体长度以及潜在的假阳性抗原(请参见结果)。根据这些分析,仅包括长度为13–23的配体,并排除了199种潜在的假阳性抗原的配体(补充材料中的表S1)。最终数据集包含来自2,604种来源蛋白的14,051个独特的配体条目。每个过滤步骤中配体条目数量的详细信息在补充材料中的图S1中显示。该数据集用作训练配体数据集,用于鉴定切割基序并生成预测配体的方法。
使用与先前配体数据相同的选择标准从研究的后期阶段从IEDB收集第二组洗脱的配体数据,并将其过滤以仅包括2016年和2017年的研究,并且这些数据不在初始配体数据集中。通过去除重复的肽来筛选数据集以消除冗余,并且也排除了没有明确提供源蛋白信息的肽。如上所述,选择了长度为13–23的配体。排除与训练数据集中具有100%同一性的任何配体匹配的配体,以及来自训练数据中源母体蛋白序列的配体。最后一组包含来自1,144个独特蛋白质序列的3,648个独特肽段。没有选择特定的限制性MHC分子,因为数据仅用于不需要了解MHC限制的验证研究。源序列是从UniProt收集的。该数据集用作评估配体数据集。
3.2 CD4 + T细胞表位数据集的收集
除了配体数据,我们收集了不同组的CD4 + T细胞表位。首先是来自我们实验室的研究中的15-mer抗原决定簇,这些抗原决定簇被T细胞识别,并以一致的格式进行了测试(称为“内部抗原决定簇”)。通过ELISPOT分析法测试了这些组中的肽在18-91个供体中的免疫识别能力,以测定以下细胞因子之一的分泌:IFN-g,IL-5,IL-10或IL-17。表S2的补充材料中列出了包括每个研究的相应参考文献在内的表位组的详细信息。在表中列出的经过同行评审的参考文献中描述了识别这些表位的详细信息。在某些情况下,基于临时分析选择了抗原决定簇,它们与发布的报告中的最终抗原决定簇列表不完全匹配。肽选择方案的简要说明如下:对于来自蒂莫西草(TG)已知过敏原的抗原决定簇,先前的研究确定了20个抗原决定簇,占对一组TG来源花粉抗原总应答的79.5%(Phl p过敏原)(TG过敏者)(27)。由于其中一些不是15聚体,因此较长的肽被“解构”以衍生出跨越这些较长的肽的15聚体。总共衍生出41个15聚体。对于来自新的蒂莫西草过敏原(NTGA)的表位,描述了19种肽包含NTGAp19肽库,选择这些肽包含针对所筛选的所有NTGA肽的总IL-5反应的至少40%(28)。对于来自屋尘螨过敏原的表位,该肽组包括34种最主要的肽,累计占筛查中检测到的总过敏原特异性反应的90%(29)。与关于TG组的描述类似,更长的区域被解构为15聚体,总共产生52个肽。对于来自蟑螂过敏原的表位,根据总SFC值> 1,000来选择71个最主要的表位(30)。对于TB抗原决定簇,我们选择了从疫苗候选抗原中鉴定出的65个15-mer抗原决定簇和捕获80%反应的先前已知抗原决定簇(31、32)。该组总共包括248个表位。源序列是从UniProt收集的。
作为第二组表位,我们使用了通过四聚体作图研究鉴定的CD4 + T细胞表位(称为“四聚体组”)。这些肽是按照以下选择标准从IEDB中收集的:“仅阳性测定,表位结构:线性序列,T细胞测定:定性结合/多聚体/四聚体(四聚体),无B细胞测定,无MHC配体测定,MHC限制类型:II类,宿主生物:智人(人类)(ID:9606,人类)。”过滤收集的数据集以仅保留可获得源抗原蛋白ID的15-mer表位。该数据集包含122个独特的表位。源序列是从UniProt收集的。
除上述表位外,我们还鉴定了五项由IEDB策划的研究,这些研究包含跨越六个蛋白质的15-mer肽,这些肽在HLA II类限制性T细胞的背景下进行了免疫原性测试。我们从这些研究中收集了表位,其中包括来自6种不同蛋白质的73个独特表位,这些蛋白完全独立于其他数据集(称为“ IEDB表位集”)(33-37)。参考资料中提供了有关此数据集收集的更多详细信息。 (38)。再次从UniProt收集源序列。
3.3 用于预测配体的“解理概率分数”的计算 Calculation of “Cleavage Probability Score” for Prediction of Ligands
我们首先使用从MHC配体数据中识别出的切割位点周围的氨基酸偏好,得出可应用于任何序列的N和C末端的“基序分数”。 首先,通过计算肽段N和C端三个氨基酸残基的频率(N-1,N0和N + 1,位于一个N端,一个N端和一个N端)的频率来完成此操作。 在N末端之后,分别对应C末端的C-1,C0和C + 1位置)。 然后将其除以整个蛋白质组中相应氨基酸的背景频率,然后对这些相对氨基酸频率进行对数转换,以计算对数比分。 对于给定的三联体氨基酸,我们现在可以通过简单地计算对数奇数得分的总和来指定一个分数,这些分数是C末端或N末端裂解:

其中FN-1,FN0和FN + 1分别是肽段N端之前一个位置,N端和N端之后一个位置的氨基酸相对频率,分别对应于C端。
接下来,我们根据肽的长度以及N-基序和C-基序得分计算出给定肽成为配体的概率。我们使用训练配体组中每种长度的肽段的相对比例作为基于长度的概率(称为长度概率)(补充材料中的表S3)。为了计算N-基序和C-基序的概率,分别根据N-基序和C-基序的分数,将整个肽组分别划分为N-基和C-基的近似相等数目的肽。然后根据相应单元中配体的数量计算出肽的N基序和C基序概率(补充材料中的表S4和S5)。然后,通过计算长度概率,N-基序概率和C-基序概率的乘积,得出每个肽的“裂解概率分数”。补充材料中的图S2显示了根据肽的长度,N-基序和C-基序得出肽的裂解概率得分的示意图。
三、结果
3.1 生成高质量的MHC配体洗脱数据集
训练配体数据集包含超过14,000种天然加工的配体,这些配体通过MS鉴定从表达MHC II类的细胞中洗脱的多肽。通过查询MHC II配体洗脱测定法从IEDB数据库中收集数据集。初始数据集包含35,367个肽段,并按照“材料和方法”部分的内容筛选了长度和明确的等位基因限制。初步筛选后,数据集包含28,007个长度为9-23的肽。在该组内,据报道作为配体的某些肽的长度分布短/长,令人担忧的是,某些肽可能富含降解产物或其他污染物,而不是来源于真正与MHC结合的肽。因此,我们着手确定某些长度的肽是否比其他长度的肽更好地与MHC结合基序相符。这是基于二项式概率分布完成的,该分布比较了配体和通过对每个配体长度改组配体残基而生成的随机肽之间的预测结合物的比例。通过改组配体中的氨基酸残基,可从配体序列中生成五组随机肽(图1B)。然后使用NetMHCIIpan-3.1预测原始配体的结合亲和力及其对应等位基因的随机肽组(12)。计算五个随机集的预测百分位数等级的中位数,并将其指定为随机肽的结合亲和力。
基于百分数等级10.0的预测结合亲和力临界值,计算了原始配体和随机肽组之间的预测结合剂数量。我们发现,在达到2.5后,不同长度的配体和改组肽之间的预测结合剂比例之间的倍数差异趋于平稳(图1C)。因此选择的最小折叠差异为2.5的长度,包括长度13-23,包含24,099个肽。最短的13个长度对应于一个9-mer结合核心和在结合核心两侧的2个侧翼残基,这与先前关于高亲和力MHC配体最小长度的实验报告一致,这表明结合亲和力下降短于12–13个残基的肽段的残留量(39,40)。值得注意的是,直到23个序列为止,长序列的折叠倍数均没有减少,这表明如此长的肽和可能更长的肽确实能够与MHC II类分子结合,这再次与先前的报道一致,即使是全长蛋白质充分变性后可结合(41)。
主要的组织相容性复合物配体洗脱测定法可能会提取未与MHC分子实际结合的肽,而是衍生自与MHC:肽复合物共洗脱的污染蛋白的降解产物,或源自MHC分子的肽他们自己。如果确实如此,则应将某些更有可能被共同洗脱的蛋白质富集为肽源,这些肽不具有与MHC结合所需的序列基序,从而将其源蛋白鉴定为潜在的假阳性抗原。使用上述相同的协议(使用二项式概率分布)来识别此类潜在的假阳性抗原,并将其从最终数据集中排除。分析确定了与随机肽相比,配体中预测结合物数量明显少的蛋白质。用于选择预测的粘合剂的百分数等级临界值为10.0。分析确定了199种蛋白质为潜在的假阳性,被排除在外(补充材料中的表S1)。此列表中一些顶级蛋白质是MHC链,例如HLA-DQα和β链,HLA-DPβ链,HLA-DMα和β链以及β-2-微球蛋白。在内质网和高尔基体网络中表达的蛋白质,例如反式高尔基体网络整合膜蛋白2和内质蛋白也在列表中。在排除了这些蛋白质并将配体的长度限制为13-23之后,数据集包含18286个配体条目。图1D显示了基于限制性HLA等位基因的配体数据分布。数据受DQ等位基因限制的配体支配。数据集中约61%的配体受四个DQ等位基因限制,而10个DR等位基因与25%的数据相关,而14%的配体受两个小鼠等位基因限制。总体而言,缺乏包含DP等位基因的数据,只有一个等位基因来自DP位点,并且在与该等位基因相关的数据集中很少有配体。
作为最后的过滤步骤,我们排除了在其源亲本蛋白质中未发现具有100%同一性的所有配体。该最终集合包括来自2,604个独特蛋白质序列的14,051个配体条目。每个过滤步骤中配体条目数量的详细信息在补充材料中的图S1中显示。图1E显示了基于配体长度的数据的最终分布。如前所述,在最后一组中,配体中最常见的长度是15,然后是14和16。长度13-17代表最终配体数据集的80%以上。总的来说,这一选择过程为我们提供了具有明确来源抗原,已知限制的MHC II类配体的数据集,并消除了多种潜在的污染物源。
3.2 MHC配体边界的氨基酸组成揭示了假定的切割motif
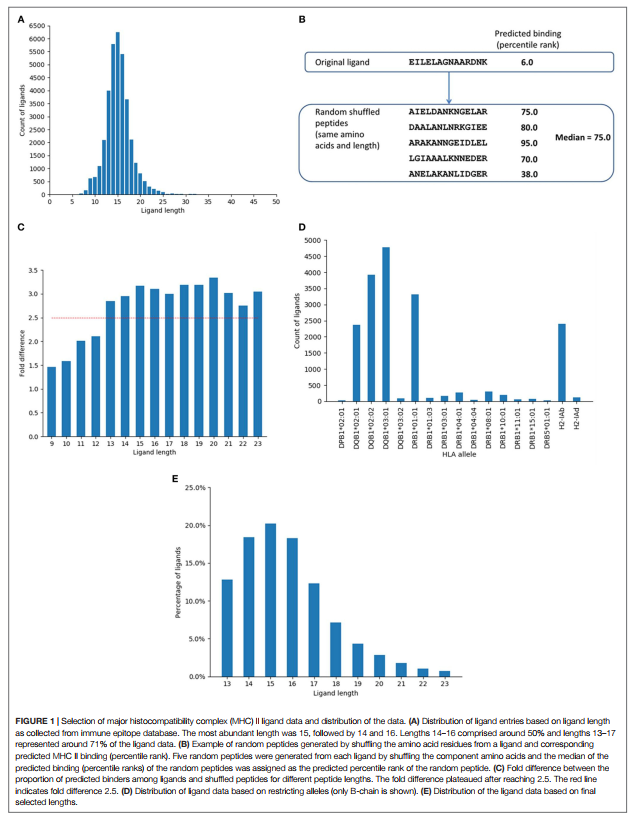
主要的组织相容性复合物配体可通过多种酶从其源蛋白上切割下来。我们假设这些酶的特异性将导致某些氨基酸在MHC配体开始和末端的切割位点比其他氨基酸更受青睐。因此,分析了配体和相邻区域的氨基酸组成。分析的序列区域在每个配体的N-和C-末端之前和之后包含10个残基,包括两个末端的残基。计算每个位置氨基酸的频率,并通过将其与整个源蛋白中的氨基酸频率进行比较来分析富集/耗竭模式。由于大多数配体的长度约为15个残基,因此从任一末端引入10个氨基酸意味着富集/耗尽基序有望重叠,而实际上它们确实重叠。图2A显示了从N末端±10位开始的位置上氨基酸的富集和耗竭。由于从C端的C-4到C-10位置显示出与从N端的N + 4到N + 10位置基本相同的基序,因此从-3到+10位置显示了C末端富集。如补充材料表S6中所示,我们着眼于与全长来源蛋白中的总氨基酸频率相比,在配体末端和相邻位置的相对频率代表的裂解基序。
由裂解基序产生的热图显示在N端的P在N0,N + 1,N + 2处的N + 1和D以及在C端的C0,I和V处的K和R在C端显着富集-1以及C0处的G,N和Q(图2A)。相比之下,热图显示了几个疏水残基的耗尽,最明显的是在C0和N + 1位置。当我们自己考虑不同的配体长度,或者仅考虑受HLA-DR或DP或DQ限制的配体时,我们检查了基序是否发生了变化。我们没有发现任何重大差异;实际上,我们生成的所有数据集的图案都非常紧密地重叠(补充材料中的表S7和S8以及图S3和S4)。在所有情况下,N和C末端的切割基序几乎都是保守的,但配体内部的基序却不同,这可能归因于与不同等位基因相关的结合基序模式的差异。这表明检测到的基序不依赖于基因座或肽长度,而是反映了抗原加工的一般偏好,这与所发现的基序与蛋白酶的众所周知的裂解基序相匹配。
我们还一致地检测到半胱氨酸残基的消耗不仅在MHC配体内部,而且还在其外部。虽然洗脱配体中半胱氨酸的耗尽可能是由于这些残基氧化和/或形成二硫键的实验问题,这将使它们更难检测,但半胱氨酸耗尽也存在于配体的侧翼区域这一事实表明:至少部分地,富含半胱氨酸的区域缺乏MHC配体的表现,这可能是由于它们形成二硫键的能力降低了它们产生MHC配体的能力(42-48)。
为了检查在MHC配体分析中发现的末端序列基序与结合MHC的肽相比是否是天然加工的,我们创建了一个对照数据集,该数据集是从我们的配体数据集的相同来源蛋白中收集的,仅包括15个如参考文献3中所述,预测使用7-等位基因方法结合MHC的Mer肽。 (49)基于推荐的7等位基因中位数共有百分等级20.0的通用阈值(作为比较,15-mer配体的平均7-等位基因共有百分位数等级为33.57。下面有〜38%的15-mer配体阈值20.0)。从该数据中产生氨基酸富集/消耗模式,方法与上面的MHC配体相同,所得热图如图2B所示,数值在补充材料的表S9A中报告。总体而言,发现从基于结合预测选择的肽获得的基序与天然洗脱的MHC配体的基序不同。虽然预期与MHC分子接触的内部位置的氨基酸偏好重叠,但是对于这些预测的结合剂,在末端没有切割基序的迹象。例如,与基于天然配体的富集模式相反,P和N在N末端的裂解位置被耗尽,而K和R在C末端被消耗。同样,在C端的K和R富集和C的耗竭也不太明显。我们使用等位基因特异性结合预测而不是7等位基因方法重复了此分析,这导致了相同的结果(补充材料中的表S9B和图S5)。这证实了MHC配体末端周围的序列基序是由于MHC结合以外的过程引起的,例如配体从其源蛋白上的裂解。
3.3 使用末端切割基序预测MHC II配体
我们想要确定我们是否可以预测性地使用配体末端的发现切割基序。在第一个分析中,我们检查了基序在训练配体数据中可以如何重新识别配体。完整的源序列被分解为长度为13-23的所有可能的肽。与配体相同的肽被认为是阳性,所有其他肽都被认为是阴性。估计了每种肽的裂解概率得分(在“材料和方法”部分中有详细信息),预计得分更高的肽更有可能成为配体。简而言之,基于肽长度和在N-和C-末端包含在N-和C-末端的三个氨基酸残基的切割基序来计算切割概率分数(肽作为配体的概率)。我们绘制了接收器的工作特性曲线(ROC曲线),并根据每种蛋白质估算了ROC曲线下的面积(AUC)。简而言之,随着分类阈值的变化以及将AUC用作预测方法性能的度量,ROC曲线将灵敏度作为二进制分类器误报率的函数进行绘制。 AUC为0.5表示完全随机预测,而AUC为1.0表示完美预测。在我们的案例中,当以每种蛋白质为基础评估性能并将AUC取平均值时,发现平均AUC为0.851。该值构成使用此评分方案的预测性能的上限,因为将图案值应用于生成它们的同一数据集。
3.4 使用独立的MHC II配体数据集验证“裂解概率分数”
为了验证切割基序预测洗脱的配体的能力,我们使用了从2016年至2017年发布的IEDB参考文献中收集的独立配体数据集,并排除了训练数据集中包含的任何配体和蛋白质序列。 该评估集包含来自1,144个独特序列的3,648个独特肽段。 我们使用基于鉴定的裂解基序得到的裂解概率分数,在评估配体数据中预测了配体。 当以每种蛋白质为基础进行评估时,平均AUC为0.767,并对AUC进行平均。
3.5 通过结合切割母题和绑定母题改进MHC II配体预测。 Improved MHC II Ligand Prediction by Combining Cleavage Motifs and Binding Motifs
我们想看看将基于切割基序的评分与MHC结合预测相结合是否会提高预测MHC II配体的整体能力。 因此,我们将基于上面得出的切割概率得分的切割基序与先前建立的“ 7-等位基因方法”结合起来,该方法用于预测与MHC II等位基因的结合(49)。 简而言之,7等位基因方法可预测肽与七个HLA-DR等位基因的结合亲和力,并计算中值预测亲和力,可作为肽与不同HLA分子混杂结合的能力的代理。 具有较低的中位数百分数等级值的肽被认为是更好的预测表位候选物。 然后将这两个分数与不同的权重合并,如下所示:
Combined score=α×cleavage probability score+(1−α)×binding score
其中,切割概率得分是基于上述切割基序的肽得分,而结合得分是来自7等位基因方法的代表MHC结合亲和力的同一肽的中位数百分等级。我们仅将15-mer配体用于序列,因为7等位基因结合预测方法仅适用于15-mer肽。裂解概率分数被转换为百分数等级,以调整为与结合分数相同的等级。参照来自相应蛋白质内的肽作为参考,对每个肽进行百分等级转换。
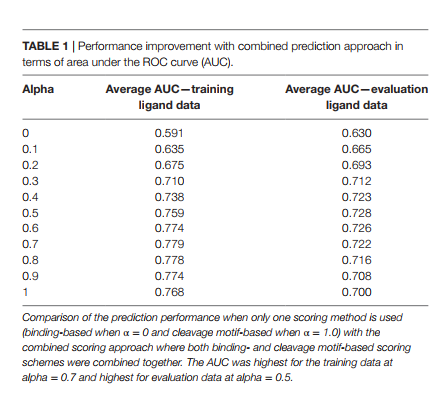
α的值从0.1到0在1到0之间变化。因此,当α= 0时,结合分数完全来自结合分数,而当α= 1时,结合分数完全由裂解概率分数赋予。总体而言,当两种评分方案都发挥作用时,而不是单独使用两种评分方法时,我们发现α值介于0.0和1.0之间时,预测性能得到了改善。该分析的结果总结在表1中。当我们在训练配体数据上应用组合评分方法时,获得的最高性能为平均AUC = 0.779(α= 0.7),这比单独的裂解概率得分略有改善(平均AUC = 0.769)。对于评估配体数据,与单独的裂解概率评分(平均AUC = 0.700)(p值= 0.02,配对t检验)相比,性能提高到平均AUC = 0.728(α= 0.5)。当使用在训练集中最佳的α值0.7时,观察到AUC = 0.722的较小改善。
3.7 切割motif无法预测CD4 + T细胞表位 Cleavage Motifs Fail to Predict CD4+ T Cell Epitopes
由于我们能够使用切割基序来预测MHC II配体,因此我们希望了解是否可以使用相同的方法来改善CD4 + T细胞表位的预测。 为了测试裂解概率评分将实际表位与其他多肽区分开的能力,我们收集了三组独立的表位:一组包含在我们实验室进行的研究中被证明可被T细胞识别的15-mer表位(“ 内部抗原决定簇”,另一个包含通过四聚体作图研究鉴定的15-mer CD4 + T细胞抗原决定簇(“四聚体集”),第三个包含由IEDB策划的对15种CD4 + T细胞抗原决定簇的抗原在6种抗原中的重叠肽的研究(“ IEDB表位组”)(有关肽组的详细信息,请参见“材料和方法”部分)。
我们在两个数据集中的T细胞表位是基于连续的15-mer肽的筛查发现的,这些肽由10个跨越其来源蛋白的残基重叠(第三个是四聚体数据集)。此类重叠的肽数据集避免了基于预测的结合MHC的能力而预选测试T细胞识别的肽的潜在偏倚。重要的是,这些肽的N-和C-末端边界是这种重叠的肽合成方案的结果,因此预期表位肽末端不是有效的切割位点。这并不妨碍其被表位特异性记忆T细胞识别,因为肽是从其源序列中释放出来的。只要测试的肽可以结合MHC分子并包含引发幼稚T细胞受体(TCR)的氨基酸序列,记忆TCR就可以识别它。因此,尽管我们的数据集中记忆T细胞识别的15-mer肽不必在其末端具有有效的切割位点,但仍需要重叠的引发肽,它们共享一个约9-mer核心与TCR结合,并且可以有效地从源蛋白中释放出来。
考虑到这些因素,我们计算了蛋白质中每个15-mer的得分,该得分评估了将与15-mer重叠的肽切割至少9个残基的能力。为此,我们首先为三个表位集合中的每个序列计算长度为13-23的所有肽的概率分数。然后,对于蛋白质中的每个15-mer肽,我们计算了与其共享9-mer结合核心的肽之间的平均得分,并将其分配给了给定的肽。与表位完全匹配的15-mer肽标记为阳性,与任何表位共有少于9个残基的15-mer标记为阴性。我们绘制了每种蛋白质的ROC曲线,并计算了三个数据集的平均AUC。我们发现所有三个数据集的AUC都在0.5左右,这表明切割概率评分未能识别出CD4 + T细胞表位。使用给定的15-mer的任何重叠肽的最高分值而不是平均分值,也得出了类似的AUC值,约为0.5,这表明我们无法检测到CD4 + T细胞表位的切割效率增加。
此外,我们想看看将切割概率分数与预测的结合分数(代表MHC结合亲和力的7等位基因方法的预测中位数百分等级)相结合是否可以改善预测性能。为此,我们使用“ 7-等位基因方法”以“中位数百分等级”(此处称为结合得分)来预测所有15个聚体的结合亲和力。与之前相同,对于每种肽,我们计算了与它共享9-mer结合核心的肽之间的平均结合得分,并将该平均结合得分分配给了给定的肽。我们首先仅使用预测的结合分数来预测内部表位组,四聚体组和IEDB表位组的表位和AUC分别为0.649、0.747和0.668。为了将这些结合分数与切割概率分数结合起来,我们首先使用与以前相同的方法,即使用α参数,将α值从0更改为1,间隔为0.1。将裂解概率分数转换为百分位数等级,以调整为与以前相同的结合分数。因此,当α= 0时,组合分数完全由单独的结合分数贡献;当α= 1时,切割概率分数归因于完整组合分数。但是我们发现任何α> 0都不能改善AUC。在所有三个数据集中,最佳性能是单独使用结合得分时。
由于使用α参数的切割基序预测和结合预测的线性组合未显示出改善,因此我们想尝试一种替代方法。为此,我们首先根据建议的中位数百分位数临界值20.0将肽类分为MHC“结合”和“非结合”两类。我们使用此“二进制”结合数据计算了AUC,内部表位组,四聚体组和IEDB表位组的AUC分别为0.538、0.596和0.582(平均值= 0.572)。此外,我们用“较差”分数(= 0.0)替换了预测的非粘合剂的裂解概率评分,而预测的粘合剂保留了我们计算的裂解概率评分(所有> 0.0)。我们用包含结合信息的更新的裂解概率分数进行了预测性能评估,发现上述表位组的AUC分别为0.537、0.601和0.555(平均值= 0.564)。总体而言,与单独使用预测结合分数时获得的相应AUC相比,组合评分方案的AUC并没有明显改善。
四、讨论区
肽与MHC分子的结合被认为是T细胞表位抗原加工和呈递途径的限制因素,但结合本身不足以产生免疫反应(50)。因此,大多数表位预测算法都依赖MHC肽结合数据进行训练。这提供了有关肽与MHC分子的“结合基序”的可靠信息,但缺乏有关如何从抗原中天然裂解肽的信息。使用从MHC II分子洗脱的14,000多种天然加工肽的数据,我们发现了抗原中天然加工配体的N端和C端周围有一个强大的“切割基序”。裂解基序在N端具有不同程度的P和D富集,在C端具有K,R,I,V,G,N和Q。同时,其他一些残基在N和C末端被耗尽,大部分是疏水残基。残基W,F,Y,I,V,L,M和C在N和C末端的不同位置以不同程度消耗。 P在N-1,N0和C + 1位置也被耗尽。该切割基序信息被用于产生评分方案,该评分方案又被用于设计预测MHC II配体的新方法。该切割基序对于MHC II类配体是通用的,而与限制性等位基因无关。
如上所述,切割基序在N端具有不同程度的P和D富集,而在C端具有K,R,I,V,G,N和Q。这使您可以了解处理本身的特殊性。首先,人们早就认识到加工可能是多种类型蛋白酶的结果(51),包括组织蛋白酶的重要作用(52)。确实,在N端D的富集与天冬氨酸蛋白酶组织蛋白酶D和E的作用相一致(53)。对P的富集与以下事实相符,即其α-氨基是仲氨基酸而不是其他氨基酸的伯氨基酸,因此不能用于从氨基肽酶降解。此外,K / R C-末端富集是胰蛋白酶样特异性的标志。实际上,胰蛋白酶早在1980年代初期就被用来定义历史上第一个II类限制性表位(54)。
除了富集与蛋白酶裂解肽末端一致的残基外,还发现在N和C末端的多个位置处耗尽了一些疏水氨基酸,并且在配体附近的半胱氨酸残基也被耗尽了。 。这可能是由于裂解基序以外的因素影响了抗原的加工和呈递(55)。具体而言,抗原的三维结构可以影响蛋白水解过程,从而控制单个配体的呈递,并确定将肽有效呈递给T细胞的速率,这对于蛋白质的疏水性核心或二硫键之间的残基来说就是这种情况。由半胱氨酸对组成(56–59)。
此外,我们不能排除使用MS鉴定天然洗脱的肽有助于检测到的某些基序。可以预期,例如,带电氨基酸的存在会影响检测肽的能力。但是,不希望这样的有利氨基酸集中在肽的末端,并且绝对不应影响其源蛋白中洗脱的配体侧翼的残基的影响。这使我们确信,直接围绕自然加工的配体的残基中检测到的基序的主要贡献者反映了切割偏好。
MHC I类和II类限制性配体的比较提供了信息。对于I类MHC,长期以来一直显示出从蛋白酶体切割,TAP运输和ER修剪开始的加工机械可形成可用于与MHC I类分子结合的配体库。但是,决定其结合I类MHC分子能力的肽的主要位置是C端残基,而同一残基则主要决定其被蛋白酶体切割和被TAP转运的能力。推测是由于共进化,这两个过程都支持相同类型的氨基酸残基,这很有意义,因为结合未提供肽的MHC分子无法有效地向T细胞展示肽(60、61)。由于负责MHC I类结合和加工的肽残基的这种重叠,将两者的结合起来预测的影响较小。相反,我们在此描述的MHC II类配体裂解基序位于负责肽与MHC II类分子结合的残基的侧翼,并且不与结合基序重叠。这就解释了为什么将MHC II类结合和裂解预测结合起来可以显着改善鉴定MHC II类配体的能力。
这项研究将切割和结合基序纳入MHC II配体的预测中,但重要的是要注意,其他几个因素也可能最终决定抗原决定簇的组成和免疫原性。例如,据信HLA-DM与MHC II类II相关的恒定链肽(CLIP)复合物相互作用,并使CLIP与MHC结合沟分离,从而影响CD4 + T细胞表位库(4,62)。抗原不同区域的相对表面可及性(主要由抗原的结构决定)也可能是整个抗原决定簇库的一个重要因素(48)。已显示与TCR接触的氨基酸组成会影响I类MHC抗原呈递的免疫原性(63),在II类MHC情况下也是如此。这些因素以及其他参数(例如通过已知TCR集合识别表位)的合并可能会进一步缩小范围(64)。
我们要强调的是,此处描述的切割基序和导出的预测方法有意保持非常简单。我们仅计算了氨基酸频率的比率即可获得C末端和N末端的切割基序。除了改变结合了两个分数的单个比例缩放参数(alpha)之外,没有执行我们的组合MHC结合和加工模型与T细胞表位数据的拟合。我们完全希望更复杂的机器学习方法能够进一步改善此处报告的性能。保持简单的目的是第一次证明MHC II类结合基序和裂解基序独立地有助于肽成为MHC II配体的可能性。
尽管切割基序具有预测MHC II配体的能力,但它不能单独或与MHC结合预测结合地预测我们表位组中的CD4 + T细胞表位。如前所述,可能还有其他几个因素影响TCRs的抗原呈递和识别。假定基于CD4 + T细胞反应性鉴定为表位的肽通常没有明确定义的末端,则可能存在基序,但位于映射的表位之外。我们在计算上考虑到这一点的尝试并未显示出在充分表征的CD4 + T细胞表位周围存在切割基序的迹象。配体中的裂解前体信号也有可能在表位数据中被稀释而无法以这种方式恢复。此外,由于我们试图以“等位基因不可知”的方式使用切割信息,即不考虑肽的HLA等位基因限制,因此该方法可能无法捕获MHC特异性信号。虽然我们尝试将MHC II类配体洗脱数据中的切割基序转化为T细胞表位预测的尝试可能不是最佳选择,但另一种可能的解释是,此类洗脱数据中富含通过抗原加工和呈递途径产生的配体,但这种情况较少常用于T细胞表位。
参考资料
- Paul, S., Karosiene, E., Dhanda, S.K., Jurtz, V., Edwards, L., Nielsen, M., Sette, A. and Peters, B. (2018). Determination of a predictive cleavage motif for eluted MHC class II ligands. Front. Immunol. 9:1795.。 网址: https://www.frontiersin.org/articles/10.3389/fimmu.2018.01795/full
