【7.2】OAS(Observed Antibody Space)抗体测序数据集
这是 Oxford Protein Informatics Group的另一个大作,该课题主的负责人为Charlotte Deane。该文发表在Journal of Immunology,IF约4.5分,
Abs是免疫系统蛋白质,可识别有害分子以进行消除。它们的序列多样性和结合多样性使得Abs成为生物制药的主要类别。最近,使用Ig基因库(Ig-seq)的新一代测序可以查询其巨大的天然多样性。然而,Ig-seq输出目前在各个寄存器中是分散的,并且往往以原始核苷酸读数的形式呈现,这意味着重复使用数据进行分析需要非常重要的努力。为了解决这个问题,我们从55项研究中收集了Ig-seq输出,覆盖了不同免疫状态,生物体(主要是人类和小鼠)和个体的超过5亿个Ab序列。我们对这些序列进行了分类,清理,注释,翻译和编号,我们整理为Observed Antibody Space(OAS)资源,并在 http://antibodiesmap.org 上提供数据。 OAS中的数据将使用新发布的Ig-seq数据集定期更新。我们相信OAS将促进免疫库的数据挖掘,以便更好地了解免疫系统和开发更好的生物治疗药物。
一、前言
抗体(或BCR)是B细胞的蛋白质产物和脊椎动物的适应性免疫的主要参与者。 它们是高度可延展的分子,几乎可以与任何Ag结合。 有机体持有多种这样的分子,增加了Ab可以识别任意Ag的可能性,从而引发免疫反应。 由于它们具有良好的延展性,它们是最着名的试剂和生物治疗剂。 继续成功利用这些分子依赖于我们辨别Ab谱系功能多样性的能力。
Ig基因库(Ig-seq)的新一代测序使研究人员能够在个体,不同生物和不同免疫状态下一次拍摄数百万个序列的快照。 对数百万Ab序列进行测序和分析的能力有可能揭示对任何Ag的免疫应答机制和免疫系统本身的功能障碍。
许多先前的研究已经解决了Ab多样性的问题,为理解人体免疫系统的动态提供了宝贵的证据。 许多分析都集中在V(D)J基因使用的频率上,这可以为创建有偏见的治疗性Ab文库提供见解。 Ab repertoire分析的另一种治疗应用是通过对抗前和外抗原攻击实验的比较纵向研究推进疫苗设计。 这些比较研究表明,不同的个体可以针对给定的疫苗收敛于相同的Ab序列。 由于测序限制,这些分析分别关注H或L链,而人们应该研究配对的文库以获得对Ab多样性更深入的了解。
测序技术的发展已经拓宽了存储和分析流程。这意味着Ig-seq研究被拆分 为各种库,导致大规模的Ab库的数据挖掘变得困难。metadata,例如亚型、年纪、课题编号等并不标准,导致提取特殊的Ab库的数据集进行比较变得困难。此外,数据通常作为原始核苷酸读数保藏。它需要非常特别的努力来将这种原始读数转换成最终决定分子结构和Ag识别的氨基酸序列。其中一些问题由提供Ig-seq特定数据集和分析管道的服务解决,例如 BT.CR wiki( https://bt.cr ),Imm-Port(http://immport.org)( 26,27),immunoSEQ Analyzer(http:// clients.adaptivebiotech.com/),iReceptor(http://ireceptor.irmacs.sfu.ca/),或VDJServer(http://vdjserver.org) 。iReceptor和VDJServer是属于自适应免疫受体库组织主要资源,为Ig-seq输出提供标准化的存储和分析管道(24)。这些服务主要致力于促进原始数据的大量存储以执行标准化测序分析。最终,由于免疫信息学不是此类服务的主要关注点,因此从这些网站下载的批量数据是有限的,将获得的原始核苷酸数据转换为适合分析的格式仍需要安装和运行其他软件包。在本研究中,我们识别,清洁,注释并将数据作为免疫诊断分析的起点。
为了解决这些问题,我们创建了Observed Antibody Space(OAS)资源,允许对Ab库进行大规模数据挖掘。 到目前为止,我们收集了55个Ig-seq实验的原始数据,覆盖了超过5亿个序列。 我们已经通过元数据组织序列,例如生物体,同种型,B细胞类型和来源,以及B细胞供体的免疫状态,以促进特定子集的批量检索以用于比较分析。 我们已将所有Ig-seq序列转化为氨基酸,同时保留与各自原始原始核苷酸序列的连接,并使用国际ImMuno-GeneTics信息系统(IMGT)方案对它们进行编号。 这些数据可用于 http://antibodiesmap 的查询或批量下载。我们相信OAS将促进数据挖掘Ab的重要性,以便更好地了解免疫系统的动态,从而更好地设计生物治疗药物。
二、材料和方法
通过文献综述获得公众可获得的Ig-seq数据集的研究登录代码列表。 大部分原始读数均来自欧洲核苷酸档案馆(European Nucleotide Archive )和国家生物技术信息中心网站(National Center for Biotechnology Information websites )。 在少数情况下,指定了另一个公共Ig-seq存储库。 从存放的数据集中手动提取元数据并以可再现的格式排列。
根据测序平台处理下载的FASTQ文件。 配对的原始Illumina read用FLASH组装。 使用FASTX-Toolkit将组装的Ab序列转换为FASTA格式。 由于Roche 454的原始读取未配对,因此使用FASTX-Toolkit将这些FASTQ文件直接转换为FASTA格式。
除非在相应的出版物中给出了这些数据,否则H链序列用同种型信息自动注释。通过使用Smith-Waterman算法将任何给定Ab序列的恒定重域1(CH1)与相应物种的IMGT同种型参考比对来进行自动同种型注释。我们为核苷酸匹配分配了2分,对于核苷酸错配或缺口分配了-1分。 IMGT同种型参考包含Ab同种型的CH1结构域的21-nt长片段。为了确保正确的同种型鉴定的高可信度,我们在Smith-Waterman算法评分函数中采用了30的保守阈值。 Smith-Waterman算法得分低于所有同种型阈值的序列被指定为“bulk”。该方案的稳健性在作者注释的Ig-seq数据集上得到证实,其中它导致99%准确的注释。大约1%的Ig-seq数据具有非常短(或缺失)的CH1结构域序列。此类序列也被指定为bulk。
IgBLASTn用于将Ab核苷酸序列的FASTA文件转化为氨基酸。然后使用IMGT方案用ANARCI解析氨基酸序列。在此步骤中,每个序列都是IMGT编号和检查符合我们的Ig折叠知识。在经典CDR和框架区或终止密码子中具有异常插入缺失的氨基酸序列被去除,因为这些被认为在结构上是不可行的。如果它的V和J基因不与基于其各自的物种氨基酸IMGT种系构建的隐马尔可夫模型对齐,则ANAR不对序列进行编号。我们还通过检测每个氨基酸序列中的重复CDR-H3区域,检查完整的序列残基注释,检查全长框架4区域,并对CDR-H3施加37个残基的长度截止值来过滤掉可能的嵌合序列。在人类,小鼠,大鼠,兔子,羊驼和恒河猴中。由于测序平台的技术限制,某些读数缺失了V区的重要部分(例如,CDR1的部分);不具有所有三个CDR的序列被丢弃为不完整的。
使用ANARCI获得OAS中可获得的V和J基因注释,其鉴定具有最高氨基酸同一性的种系基因。 由于骆驼的V和J基因尚未很好地表征,我们在骆驼Ig-seq数据查询中使用羊驼(最接近的相对可用)Ig基因,因为这两个物种属于同一生物家族(骆驼科)。 如果将来自其他编目较差的物种的数据添加到OAS,我们将使用最接近的可用相对于V和J基因注释。
使用上述方案,我们注释了55项独立研究的Ig-seq结果。 为了简化使用新数据更新OAS的过程,我们生成了一个从原始序列读取归档中自动识别Ig-seq数据集的过程。 我们将Ab注释协议应用于保存在国家生物技术信息中心/欧洲核苷酸档案库中的每个原始核苷酸数据集; 如果我们在任何给定的数据集中找到超过10,000个Ab序列,则将其留作手动检查。 为了有效地分配元数据,仍然需要手动检查,因为这些元数据当前以非标准化的方式存放。 该程序允许自动识别新的Ig-seq数据集并半自动更新OAS。
三、结果
我们从55个Ig-seq研究中收集了原始测序输出。 使用IgBLASTn将所有原始核苷酸读数转化为氨基酸。 在OAS内,可以从翻译的氨基酸序列连接回原始核苷酸数据。 然后使用ANARCI对完整的氨基酸序列进行IMGT编号。 除了提供IMGT和基因注释外,ANARCI还可作为可能错误的Ab序列的粗刷过滤器(参见材料和方法)。 对于每个Ig-seq数据集,我们提供从IgBLASTn输出以及ANARCI解析后检索的氨基酸总数。 这些数字可用作数据集质量评估的代理。 对所有序列应用相同的检索,氨基酸转换,基因注释和编号方案可确保55个异源Ig-seq数据集中的相同参考点。 该协议产生完整的IMGT编号序列以及55个数据集中每一个的基因注释。
每个数据集中编号的氨基酸序列按元数据(例如,个体,年龄,疫苗接种方案,B细胞类型和来源等)分类(图1)。 此类元数据的存在目前尚未标准化,并且需要对每个数据集进行临时手动管理。 为了使用这样的元数据组织Ab序列,我们将每个数据集中的序列分组为数据单元。 每个数据单元表示给定数据集中的一组序列,其具有元数据值的唯一组合。 元数据值总结在表I中。
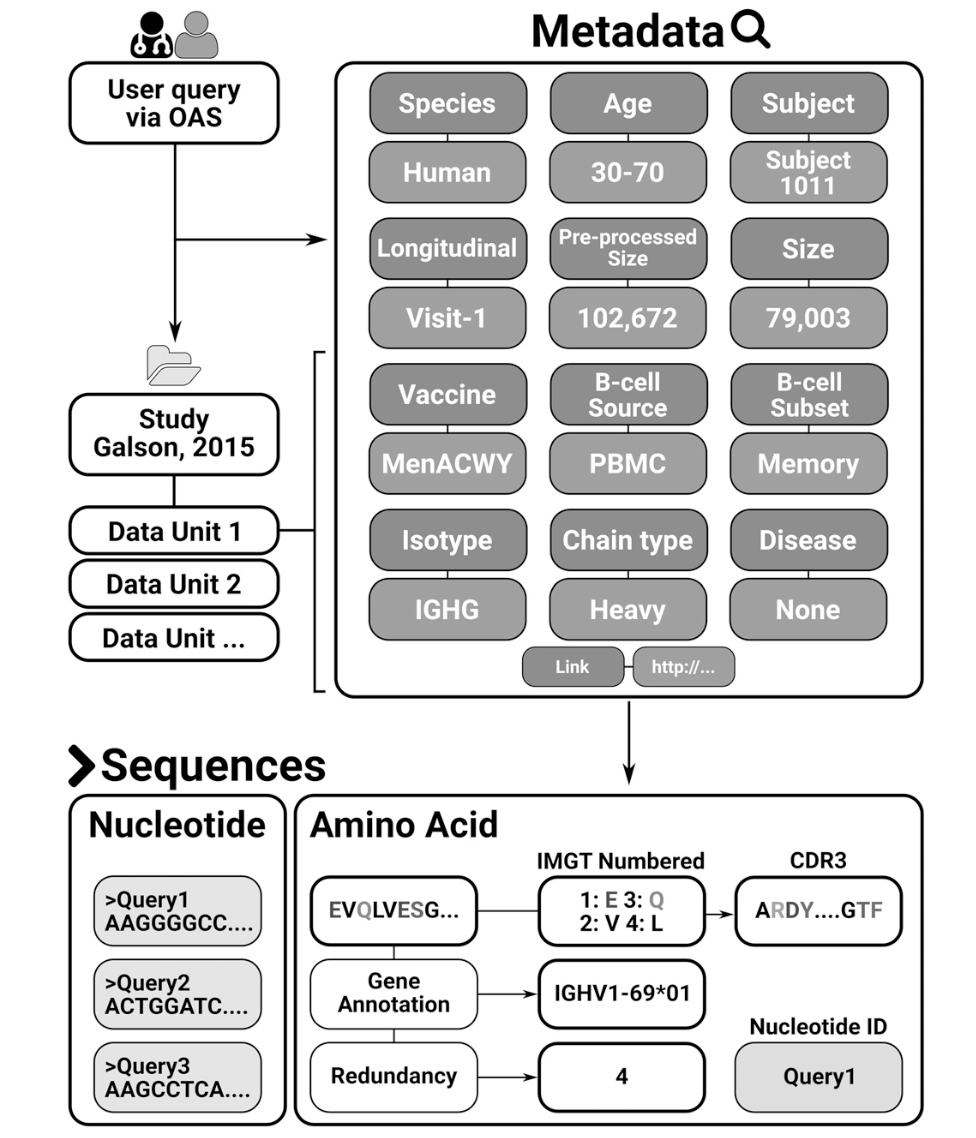
图1. OAS数据库。 来自55项研究的数据被分类为数据单位。 每个数据单元是一组共享同一组元数据的Ab序列。 数据单元中的每个序列还与序列特定的注释相关联。

截至2018年7月1日,55个Ig-seq研究纳入OAS,共计618,371,034个序列(562,544,071 VH和53,526,963个VL序列),而在ANARCI解析之前从IgBLASTn输出获得的翻译氨基酸序列总数为803,508,673 。存储在OAS中的大多数序列是鼠(~49.4%)和人(~48.4%)。 22例Ig-seq研究询问患病个体的免疫系统,最常见的疾病是HIV(13项研究)。该数据库还包含24个 naive Ab基因库的Ig-seq研究(来自捐赠者的B细胞的集合,这些B细胞是健康的并且没有有目的地接种疫苗)。 OAS数据库中B细胞的主要来源是外周血(~241万个序列),其次是脾/脾细胞(~198万)和骨髓(~124万)。该数据库保存每个重链序列的同种型信息,两种最常见的同种型是IgM(~316 mln)和IgG(~144 mln)。对于约6500万个序列,我们无法高可信度地分配同种型。 OAS数据库中Ig-seq研究的中位冗余大小为2,164,901个序列,而最大的Ig-seq研究是Greiff等人的研究(246,449,120个冗余序列)。如果它们具有相同的长度和相同的氨基酸组成,则两个序列是多余的。表II中给出了每个数据集的详细统计数据,摘要统计数据位于 http:// antibodymap.org/oasstats 。所有数据都可以批量下载或在 http://antibodymap.org 查询单个数据单位。
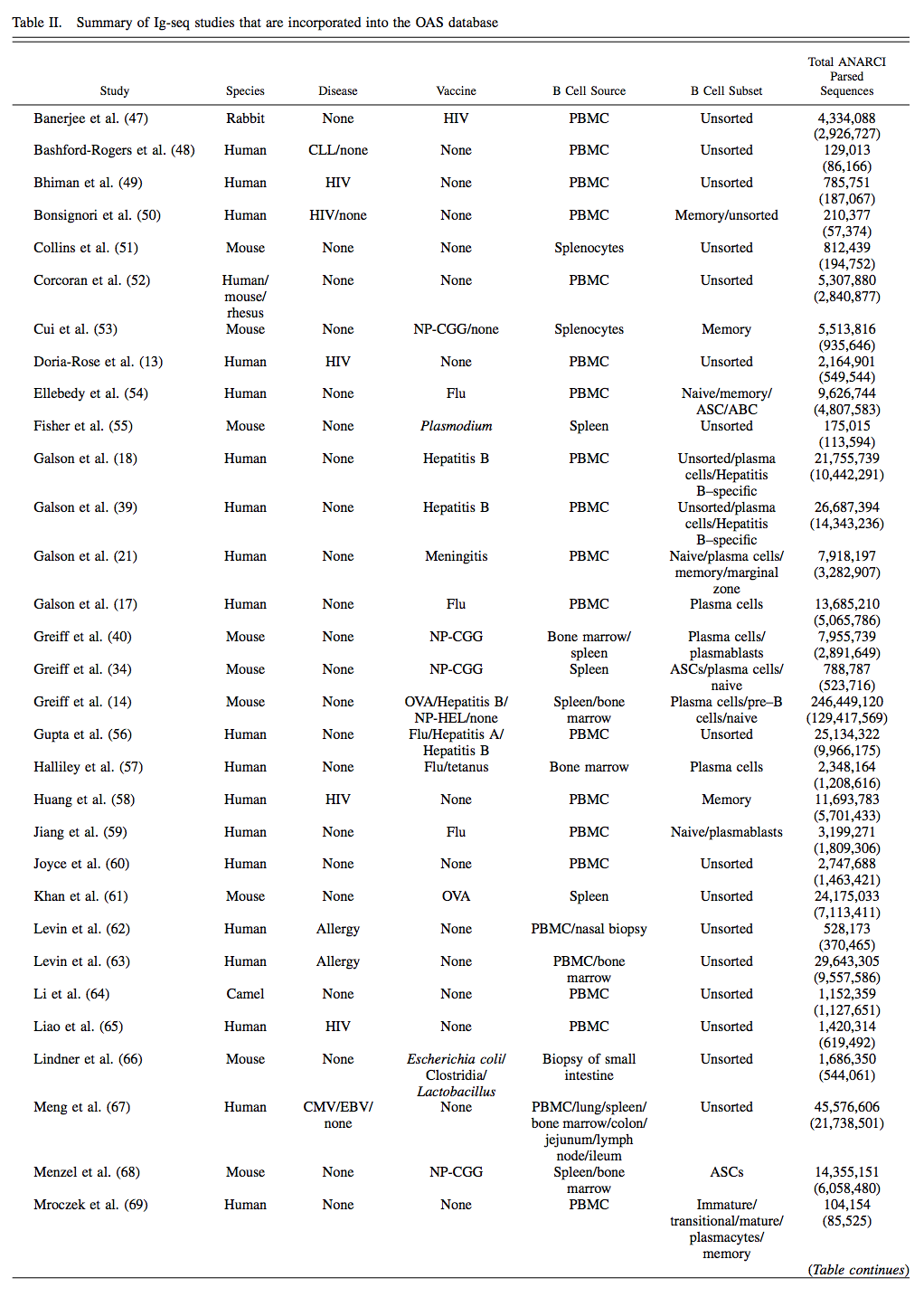
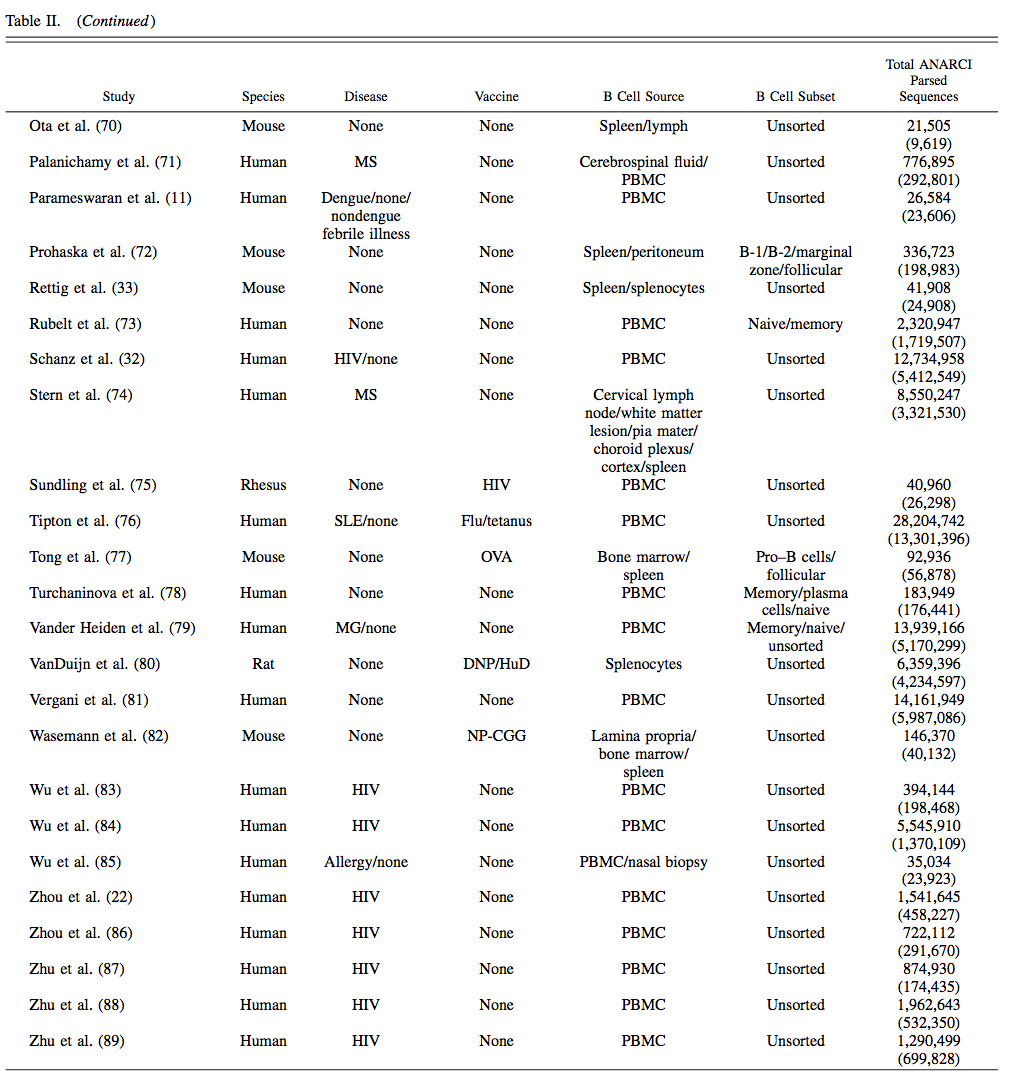
将数据集组织成与给定Ig-seq实验相关的研究。 OAS数据库中的每项研究都细分为数据单元。 每个数据单元是由表I中给出的元数据描述符唯一识别的IMGT编号的氨基酸序列的集合; 其中五种(物种,疾病,疫苗,B细胞来源和B细胞类型)在此表中给出。 “Total ANARCI Parsed Sequences”列表示数据库中冗余序列的总数,括号中的非冗余数字。
ABC,激活B细胞; ASC,Ab分泌细胞; CLL,慢性淋巴细胞白血病; DNP,二硝基苯修饰的匙孔血蓝蛋白; Flu,流感; HuD,副肿瘤性脑脊髓炎Ag; MG,重症肌无力; MS,多发性硬化; NP-CGG,鸡g球蛋白; NP-HEL,鸡蛋溶菌酶; SLE,系统性红斑狼疮。
四、讨论
在本研究中,我们描述了OAS数据库,这是一个统一的存储库,用于促进Ab谱系的氨基酸和核苷酸形式的大规模数据挖掘。在Ig-seq存储空间中缺乏完善的储存库需要我们将数据集的文献检索和手动管理相结合,以将数据组织到OAS中。目前缺乏广泛采用的存储标准阻碍了OAS的自动更新,因为我们发现大量Abs的数据集仍需要手动策划才能正确执行元数据注释。希望,自适应免疫受体库群体的努力将导致Ig-seq输出的标准化,并将进一步简化存储程序,促进Ab谱系的大规模数据挖掘。由于数据集的大小以及生物信息学家,湿实验室科学家和临床医生所需的各种数据描述符和分析管道,设计统一的Ab谱系库存具有挑战性。
据我们所知,OAS是第一个有组织的大量Ig-seq输出的集合,随着越来越多的Ig-seq数据可用而不断扩展。基本数据文件通过轻量级元数据条目,以高效压缩格式存储并可搜索。 为了在不同的Ab库的子集中进行比较生物信息学分析,我们通过常用的元数据描述来注释数据集,例如生物体,同种型,B细胞类型和来源,以及B细胞供体的免疫状态。 为了促进关于特定Ab序列或区域的研究,我们制备完整的IMGT编号的高质量氨基酸序列以及基因注释以及连接的原始核苷酸数据。
这些数据应有助于跨不同研究的深入比较分析,以辨别独立样本之间观察到的共性,以及指导Ig-seq实验尚未被查询的Ab库。揭示共享偏好在识别战略上用于启动免疫反应的理论上允许的Ab空间的部分方面是非常宝贵的。此外,这种比较研究可以提供一种解除免疫谱的各种df的方法,例如同种型或生物的多样性之间的差异。绘制人/小鼠库之间的差异对于设计更好的人源化生物治疗剂特别有意义。配对的Ab链序列信息提供了对Ab生物学的增强观点。然而,目前的配对测序方法仅允许描绘CDR-H3和CDR-L3序列;随着测序阅读长度增加以跨越所有三个CDR区域,这些配对的Ig-seq数据集将被整合到OAS中。
除了确定整个库的广泛共性之外,数据挖掘Ig-seq输出为设计更好的基于Ab的疗法提供了新的途径。过多的目前可用的Ig-seq数据提供了一组应该能够在生物体中折叠和起作用的序列的一瞥。将治疗候选物与Ig-seq谱系中的序列对齐可揭示可能自然可接受的突变选择,因此提供Ab工程的捷径,例如人源化。此外,将自然观察到的Abs与治疗性腹肌进行对比可以提供关于这些分子的自然有利的生物物理特性的见解。所有这些未来的应用都依赖于结构良好的数据集的可用性,这些数据集可以为生物信息学分析提供统一的参考点。我们希望OAS将帮助数据挖掘Ab的所有组成部分,帮助确定我们的免疫系统的战略偏好,并最终改善我们如何将Abs设计成更好的治疗方法。
参考资料
- September 14, 2018, www.jimmunol.org/cgi/doi/10.4049/jimmunol.1800708 . Observed Antibody Space: A Resource for Data Mining Next-Generation Sequencing of Antibody Repertoires
