【7.8.2.2】FGF(FGFR)
FGFR(fibroblast growth factor receiptor,成纤维细胞生长因子)
打破肿瘤微环境的保护壁垒,是抗肿瘤药物开发的重要方向。FGFR抑制剂,近3年连续获FDA批准3个新药,获批适应症均为临床急需,迎来了开发道路上的多个里程碑。而从泛抑制剂到单一亚型药物的开发,临床在研品种十余个,又可见其潜力巨大。不难看出,FGFR绝对为当下肿瘤领域药物开发极热的靶点之一!
一、FGFR/靶点重要信息
肿瘤微环境重要的组成之一,即为肿瘤相关成纤维细胞;其与正常纤维细胞相比,是体积较大的纺锤形间充质细胞;活性受肿瘤细胞分泌的生长因子调控;同时,其自身还可以分泌成纤维细胞生长因子,即FGF。
成纤维生长因子(fibroblast growth factor,FGF)家族:由22个配体组成,其中18个配体通过4个高度保守的跨膜酪氨酸激酶受体FGFR1-4发挥作用,配体与受体结合可促进受体二聚化,激活下游信号传导通路,如PLC-γ/Ca2+、RAS/MAPK、FRS2/PI3K/AKT及PLC-γ/PKC等通路。这些信号传导途径在多种生理过程如细胞增殖、分化、迁移和凋亡中均起着至关重要的作用。
分布方面:在鳞状非小细胞肺癌、乳腺癌及食道癌中观察到了FGFR1的扩增,在胃癌和乳腺癌中发现了FGFR2的扩增,在膀胱癌、子宫内膜癌和肺鳞状细胞癌中观察到FGFR的激活点突变,在多发性骨髓瘤中观察到易位、FGFR3的扩增和突变。

1. 靶点机制(图 42)
肿瘤微环境重要的组成之一是肿瘤相关成纤维细胞;与正常纤维细胞相比,是肿瘤相 关成纤维细胞是体积较大的纺锤形间充质细胞;活性受肿瘤细胞分泌的生长因子调控;同 时,其自身还可以分泌成纤维细胞生长因子(fibroblast growth factor,FGF)。FGF 作为 FGFR 的配体家族,由 22 个功能不同的配体组成,其中 18 个配体通过 4 个高度保守的跨 膜酪氨酸激酶受体 FGFR1-4 发挥作用,配体与受体结合可促进受体二聚化,激活下游信 号传导通路,如 PLC-γ/Ca2+、RAS/MAPK、FRS2/PI3K/AKT 及 PLC-γ/PKC 等通路。这些 信号传导途径在多种生理过程如细胞增殖、分化、迁移和凋亡中均起着至关重要的作用。 FGFR 在多种细胞类型上表达。当 FGFR 发生突变或过表达时,会引起 FGFR 信号通路过 度激活,并进一步诱发正常细胞癌变。
研究发现,多种肿瘤的发生中伴随着肿瘤组织的 FGFR 过表达和激活,它们可促进肿 瘤血管形成和肿瘤细胞分裂增殖等。FGFR 作为受体酪氨酸激酶超家族的一员,几乎在各 种恶性肿瘤中均存在不同程度的异常,发生率较高的恶性肿瘤有尿路上皮癌、胆管癌、乳 腺癌、子宫内膜癌和鳞状上皮癌等;同时,在肺癌、肝癌及乳腺癌等肿瘤中也发现了 FGFR 的异常激活。因此,FGFR 已经成为全球制药公司开发新型抗肿瘤药物的重要靶标之一, 引起各国药物学家广泛关注。
2. 临床研究申报概况
目前,国内进入临床研究阶段的有 24 项(有 2 项是国际多中心试验),已完成 3 项, 21 项正在进行中。这些产品大致可分为泛(多靶点)FGFR 抑制剂和选择性 FGFR 抑制 剂。泛 FGFR 亚型的研究药物较多共 9 款,包括仑伐替尼、瑞戈非尼、帕唑帕尼、Gunagratinib、 法米替尼、MAX-40279、安罗替尼、FH-2001、CGT-6321。选择性 FGFR 药物共 4 款,为SYHX2005、Fexagratinib、索凡替尼、ABSK061。其中,进入III期临床试验阶段的药物有 1 款,为安罗替尼。进入II期临床试验阶段的药物有 4 款,为 Fexagratinib、Gunagratinib、 索凡替尼、MAX-40279。进入I期临床试验阶段的药物有 6 款,为 SYHX2005、法米替尼、 ABSK061、MAX-40279、FH-2001、CGT-6321。有 3 款处于 BE 阶段的药物,为仑伐替 尼、瑞戈非尼、帕唑帕尼。
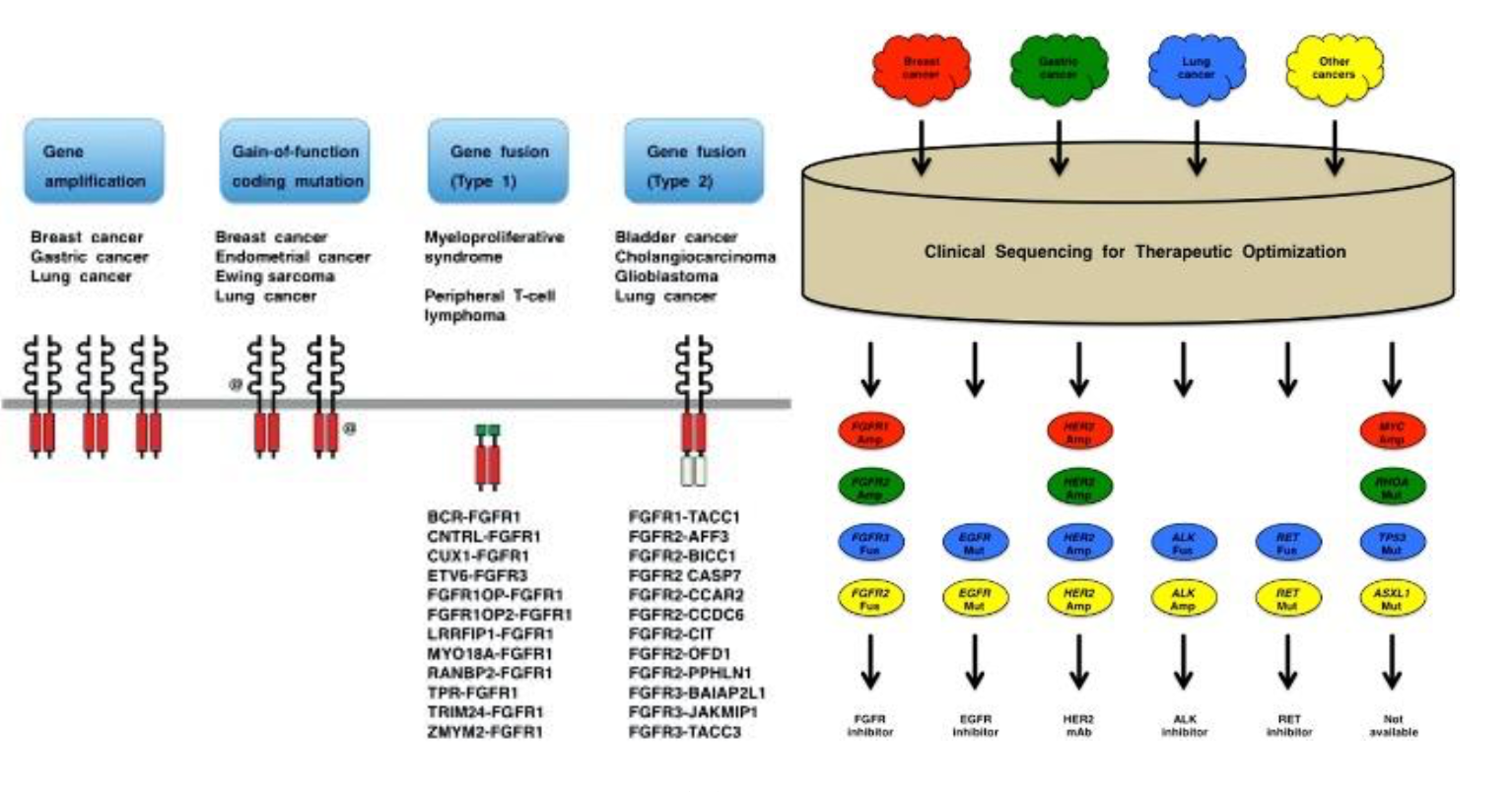
资料来源:Katoh M.FGFR inhibitors:efects on cancer cells,tumor microenvironment and whole-body homeostasis(Review)[J].Int J Mol Med,2016,38(1):3-15.doi:10.3892/ijmm.2016.2620.Epub 2016 May 31.PMID:27245147; PMCID:PMC4899036.
抗 FGFR 或 FGF 是针对于有 FGF 和/或 FGFR 改变肿瘤的一种有前景的治疗方法。 2019 年 4 月,Erdafitinib 被加速批准用于 FGFR 改变的尿路上皮癌,pemigatinib 在 2020 年 4 月被加速批准用于 FGFR2 融合或其他重排的胆管癌。FGF-FGFR 信号通路受到了更多 的关注。针对这一途径的药物,包括非选择性和选择性 FGFR TKI、抗 FGF/FGFR 单克隆 抗体和 FGF 配体陷阱。另外, 选择性FGFR抑制剂又可分为非共价FGFR抑制剂和共价FGFR 抑制剂。由于泛 FGFR抑制剂的全身毒性,更多的选择性非共价 FGFR 抑制剂被开发出来。尽管非选择性 TKI 药物 Pazopanib 和 Ponatinib 在伴有 FGFR2 融合的肝内胆管癌 (intrahepatic Cholangiocarcinoma,ICCA)患者中显示出了抗肿瘤活性,但其他临床前和临 床试验强调了使用非选择性 FGFR-靶向治疗药物的缺陷,包括脱靶副作用的问题。因此, 使用选择性 FGFR 激酶抑制剂是解决这些问题的合理方法。
3. 简评
过去 10 年对 FGF 及其相关的 FGFR 进行了广泛研究,研究重点是 FGF-FGFR 信号 的治疗潜力。FGF 通路由 22 个人类 FGF 和 4 个高度保守的跨膜受体组成,即 FGFR1-4, 具有细胞内酪氨酸激酶结构域,由 4 种独立基因编码,通过硫酸乙酰肝素或 Klotho 依赖 性途径介导 FGF 信号转导。FGFR 是单链糖蛋白,由胞外区、跨膜区和胞内区组成。在细胞表面结合 FGF 后,配体依赖的受体二聚体触发 FGF-FGFR 信号通路。这导致受体激酶 结构域的细胞内磷酸化,细胞内信号的级联,以及激活许多细胞内生存和增殖途径的基因 转录。FGF-FGFR 相互作用的特异性受到受体副核苷酸的不同配体结合能力、FGFR 的选 择性剪接、配体和受体的组织特异性表达,以及促进 FGF-FGFR 相互作用并增加配体特 异性的细胞表面或分泌蛋白的影响。虽然 FGFR 抑制剂的疗效令人鼓舞,但其反应率和反 应持续时间低于传统上在其他癌基因成瘾肿瘤(如 EGFR 或 ALK 驱动的肺癌)中观察到 的疗效。虽然多克隆 FGFR2 激酶结构域突变形式的获得性耐药缩短了患者受益的持续时 间,但连续活检和 ctDNA 分析可以帮助确定耐药机制,并指导 FGFR 抑制剂的后续使用。 总之,为了扩大和延长 FGFR 抑制剂对患者的益处,我们需要更好地了解其原发性和继发 性耐药机制,并针对其机制开发可以延迟或克服耐药的联合抑制剂和下一代抑制剂。
参考资料
- https://mp.weixin.qq.com/s/FICl5JPbgwOdFGUCjGfAng
- 《2022年度中国抗肿瘤新药临床研究评述》
