【3.7.1】肠炎性肠道疾病中肠道微生物生态系统的多组学
数据下载:https://ibdmdb.org/tunnel/public/summary.html
摘要:
包括克罗恩氏病和溃疡性结肠炎在内的炎症性肠病影响全球数百万人。克罗恩氏病和溃疡性结肠炎是复杂的疾病,在临床,免疫,分子,遗传和微生物水平上是异质的。个体的影响因素一直是广泛研究的重点。作为整合型人类微生物组计划(HMP2或iHMP)的一部分,我们每132名受试者进行了为期一年的随访,以生成疾病期间宿主和微生物活动的整合纵向分子分布图(每个长达24个时间点;总共2,965个粪便,活检,和血液标本)。在这里,我们介绍了结果,这些结果提供了炎症性肠病活动期间肠道微生物组功能失调的全面视图。我们证明了:
- 兼性厌氧菌的特征性增加(以专性厌氧菌为代价),
- 以及微生物转录(例如,梭状芽孢杆菌),
- 代谢产物池(酰基肉碱,胆汁酸和短链脂肪酸)的分子破坏
- 宿主血清中的抗体。
疾病活动的时期还以时间变异性增加为特征,具有特征性的分类学,功能和生化变化。最后,综合分析确定了这种失调的关键微生物,生化和宿主因素。该研究的基础设施资源,结果和数据可通过炎症性肠病多组学数据库( http://ibdmdb.org )获得,提供了有关炎症性肠病中宿主和微生物活动日期的最全面描述。
一、主要内容
炎症性肠病(IBD,Inflammatory bowel diseases)影响超过350万人,其发病率在全球范围内正在增加1。这些疾病最普遍的形式是克罗恩病(CD,Crohn’s disease)和溃疡性结肠炎(UC, ulcerative colitis),其特征是胃肠道(对于CD)或结肠(在UC中)使人衰弱和慢性复发并缓解炎症。这些条件下从主机之间的复杂相互作用导致2,3,微生物4,5,6,和环境7分的因素。人类基因组中IBD的驱动因素包括200多种风险变异,其中许多与宿主-微生物相互作用有关3。IBD个体肠道微生物组的常见变化包括兼性厌氧菌(包括大肠杆菌8)的增加,以及专性厌氧短链脂肪酸(SCFA)的产生者4的减少。在这里,为了支持对IBD相关的肠道微生物组的病因学的系统级了解,而不仅仅是先前报道的宏基因组学概况,我们将IBDMDB作为人类综合微生物组计划的一部分进行介绍。
我们从五个学术医疗中心招募了 132 名参与者(三个儿科亚组:辛辛那提儿童医院、马萨诸塞州总医院 (MGH) 儿科和埃默里大学医院;以及两个成人组:MGH 和 Cedars-Sinai 医疗中心;图1a、扩展数据表1,见 方法)。根据最初的内窥镜和组织病理学发现未被诊断为 IBD 的个体被归类为“非 IBD”对照。我们分析了在诊所收集的 651 份活组织检查(基线)和 529 份血液样本(大约每季度一次),以及 1,785 份粪便样本,这些样本在一年内使用家庭运输协议每两周收集一次(图1b))。后者在所有受试者的几个“全局”时间点产生主要以微生物为重点的特征:宏基因组(MGX)、宏转录组(MTX)、蛋白质组(MPX)、代谢组(MBX)和病毒组(VX)(图1b),如以及从具有更多可变疾病活动的个体中进行更密集、更密集的采样(参见 方法,扩展数据图1a-d)。我们从许多单独的粪便样本中生成了多种测量类型,包括产生所有粪便衍生测量值的 305 个样本,以及 791 个 MGX-MTX 对(图1c,扩展数据图1b))。活检产生了宿主和微生物靶向的人类 RNA 测序(RNA-seq (HTX))、表观遗传减少代表性亚硫酸氢盐测序(RRBS)和 16S rRNA 基因扩增子测序(16S),它们与人类外显子组测序、血清学谱、和来自血液的 RRBS。所有数据均可在 https://ibdmdb.org/ 获得。
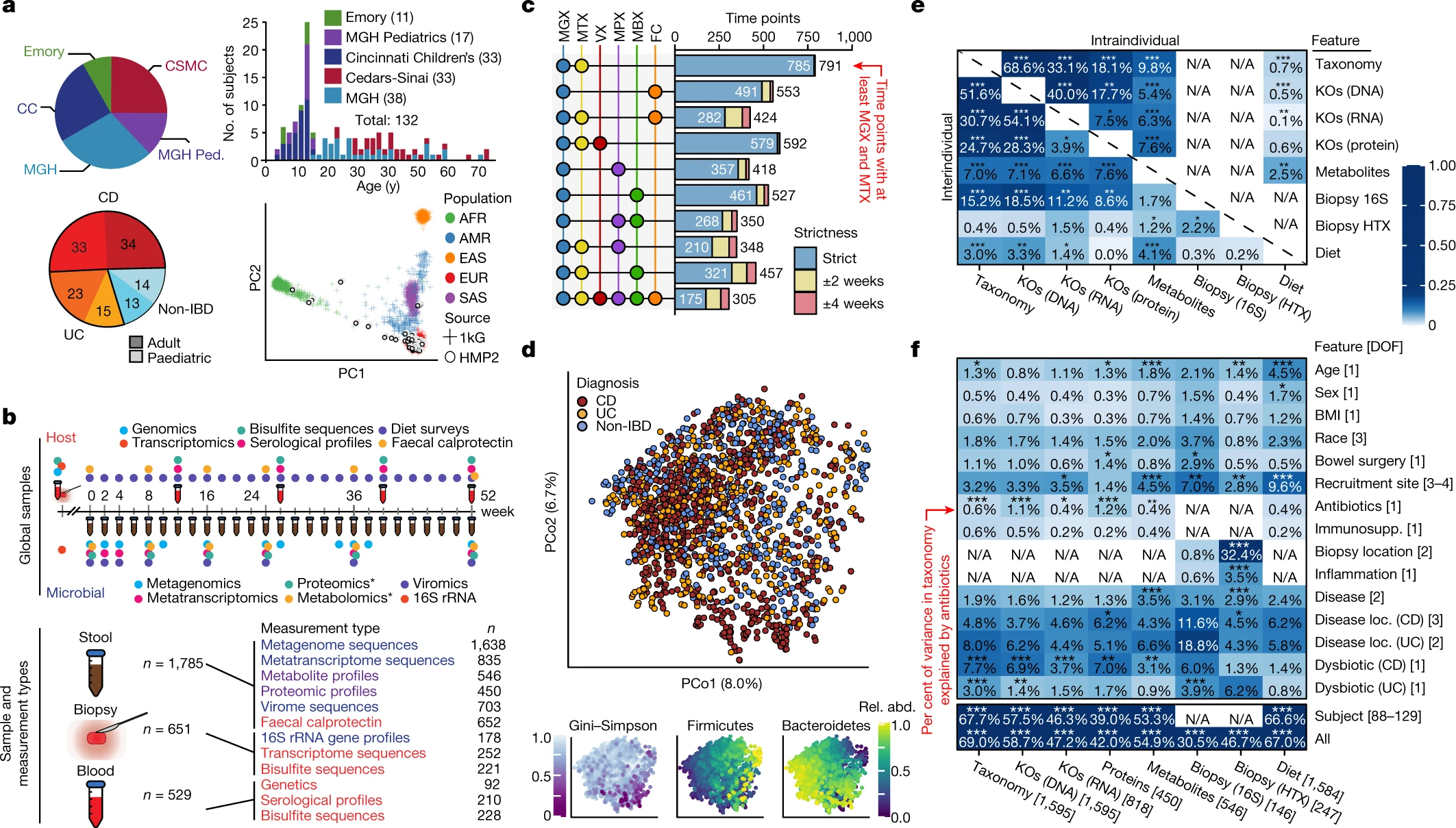
a,队列特征概述。我们对 132 名参与者(有 CD、有 UC 或没有 IBD(对照))每人进行了一年的随访。SNP 配置文件的主成分分析 (PCA) 显示,与 1000 基因组 (1kG) 参考相比,生成的 IBDMDB 队列主要是欧洲血统(参见 方法)。b、抽样策略。该研究从结肠活检(基线)、血液(大约每季度一次)和粪便(每两周一次)中产生宿主和微生物数据,评估所有受试者的全球时间点和一个子集的密集时间课程。显示了原始的、非质量控制的样品计数。C, 来自同一样本(严格)或接近一致的时间点(差异长达 2 或 4 周;参见方法)的多组学测量的重叠 。d,基于物种级 Bray-Curtis 相异性的主坐标分析 (PCoA);大多数变异是由拟杆菌门与厚壁菌门之间的权衡驱动的。来自 IBD 个体(尤其是 CD)的样本的 Gini-Simpson α 多样性略低( 与非 IBD 相比,UC 和 CD 的Wald 检验P = 0.26 和 0.014,分别)。e,Mantel 测试量化测量类型对之间解释的方差(Mantel 统计量的平方),具有跨受试者(个体间)或受试者内随时间(个体内部;见 方法); 结果显示了测量类型之间的紧密耦合。f 中的样本大小。f,PERMANOVA 显示所有测量类型的个体间变异最大,即使是相对较大的影响(例如,抗生素或 IBD 表型)也捕获较少的变异(参见 方法)。分层测试 (CD/UC) 仅考虑指定表型内的样本(请注意,这些样本的样本数会减少,从而偶然导致更大的预期协变)。星星显示 FDR 校正的统计显着性(FDR * P ≤ 0.05,** P ≤ 0.01,*** P ≤ 0.001)。独立地估计每个特征的方差(方法)。“全部”是指具有所有元数据的模型。每种测量类型的总n显示在方括号中,分布在多达 132 名受试者中(扩展数据图1a,参见 方法)。
二、IBD 中的多组学肠道微生物组变化
与先前的研究4、5一致,尽管 IBD 的子集(特别是 CD)对基于分类的主坐标的第二个轴做出了贡献(图1d,扩展数据图2a),但个体间的变异占方差的大部分对于所有测量类型5,9,10(图1E中,f,扩展数据图2a中)。即使是相对较大的影响,如疾病状态或生理和技术因素,也解释了较小比例的变异(图1f);这在所有测量类型中都是正确的,尽管这些捕获了 IBD 生态失调的不同方面(见下文)。
大多数测量类型在横截面和纵向上捕获了受试者之间和内部的相关变化(图1e)。从 MGX、MTX 和 MPX 测量的功能配置文件是最紧密耦合的(图1e),尽管一些单独的特征相关性很弱(Spearman 相关性 MGX-MTX 0.44 ± 0.10(平均值 ± sd),MGX-MPX 0.14 ± 0.083,和 MTX–MPX 0.18 ± 0.096l;扩展数据图2b)。出乎意料的是,被表征的酶往往与其已知的底物或产物只有微弱的相关性(补充图1)。虽然我们的饮食特征是通过一个非常广泛的食物频率问卷获得的,但它提供了大量人口中纵向饮食-微生物组耦合的初步特征。饮食占受试者之间分类变异的3%(错误发现率 (FDR) P = 7.4 × 10 -4)和纵向变异的0.7%(FDR P = 4.3 × 10 -4)。
IBD 个体与非 IBD 个体之间的简单横截面差异(补充表 1-14)在代谢组中最为明显(图 1f、2a,扩展数据图 2a、c、d,参见方法)。总体而言,IBD 患者的代谢物池多样性较低,与之前对微生物多样性的观察结果相似(补充表2);这可能是由于营养吸收不良、肠内水分或血液含量较高以及活动性 IBD 患者的肠道转运时间较短所致11. 在 IBD 患者中含量较多的较少数量的化合物包括多不饱和脂肪酸,如肾上腺素和花生四烯酸。 泛酸和烟酸(分别为维生素 B5 和 B3)在 IBD 期间在肠道中特别消耗;这是值得注意的,因为这些通常不属于 IBD 12患者血清中缺乏的 B 族维生素,尽管在活动性 CD 13期间检测到低烟酸水平。两种维生素都需要产生用于脂质代谢的辅助因子14,而烟酸盐在肠道15 中具有抗炎和抗凋亡功能。值得注意的是,烟酸,烟酸16的代谢物,几乎完全在 IBD 患者的粪便中被发现。粪便钙卫蛋白和 Harvey-Bradshaw 指数(HBI)是 CD 疾病严重程度的两个指标,没有显示出显着相关性,而UC 中的简单临床结肠炎活动指数17(SCCAI)确实与粪便钙卫蛋白水平弱相关(图2b) .
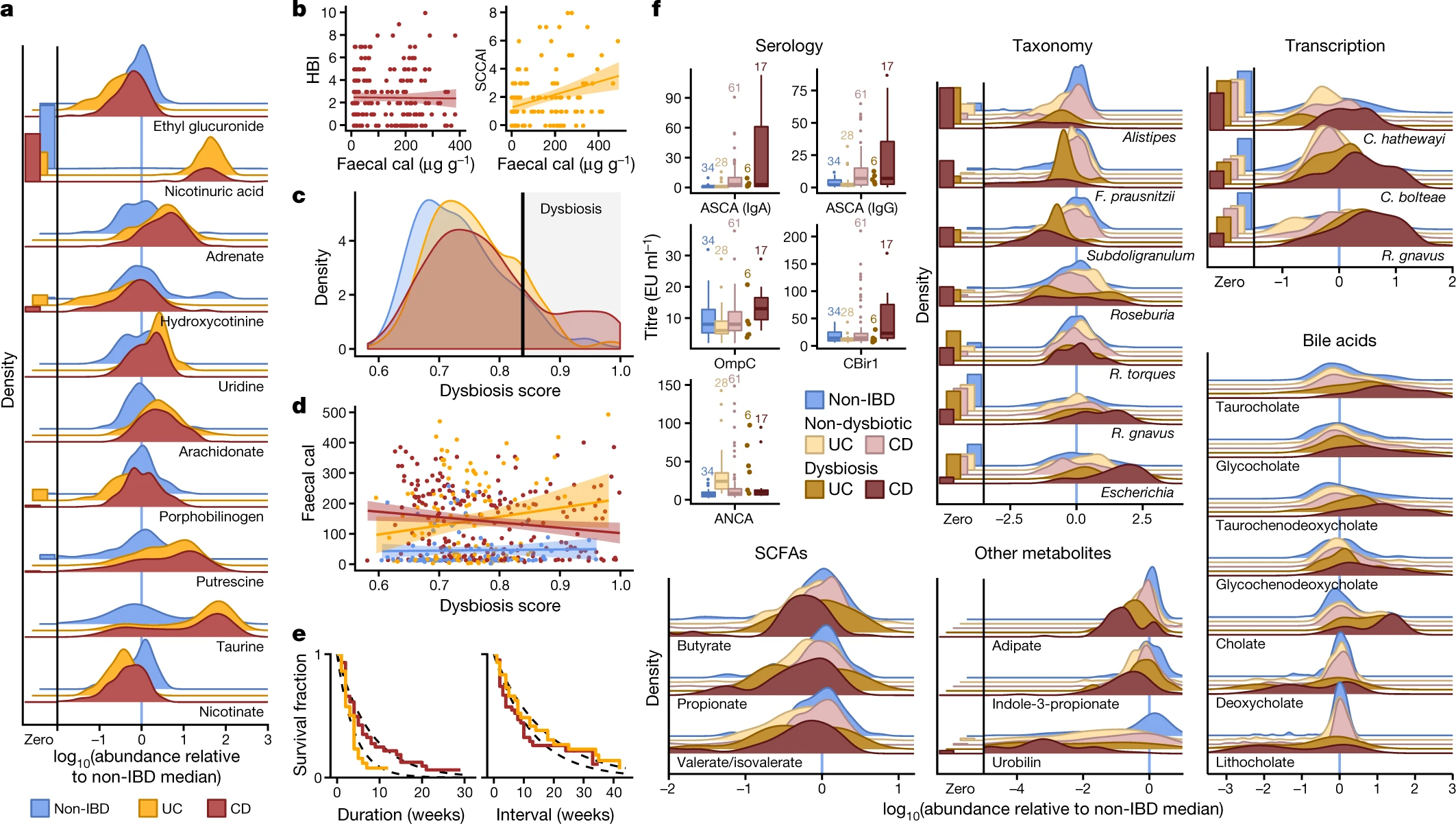
a,IBD 个体样本中 10 种最具横断面显着差异丰度的代谢物的相对丰度分布,作为与非 IBD 个体的中位相对丰度的比值(Wald 检验;所有 FDR P < 0.003;参见 方法;补充)表1 – 14)。左图,低于检测限的样品比例(参见 方法)。n = 106 名受试者的 546 个样本。b,两种疾病活动度测量之间的关系:患者报告(CD 中的 Harvey-Bradshaw 指数(HBI),n = 65 名受试者的 680 个样本;UC 中的简单临床结肠炎活动指数(SCCAI),n) = 来自 38 名受试者的 429 个样本)和宿主分子(粪便钙卫蛋白 (cal) 43,n = 来自 98 名受试者的 652 个样本)。线性回归显示为 95% 置信区间。c , d,作为疾病活动量度的微生物失调评分分布(c,样本和非 IBD 样本之间的中值 Bray-Curtis 差异;参见 方法)及其与钙卫蛋白的关系(d,n = 98 名受试者的 652 个样本)。95% 置信度的线性回归。电子, UC 和 CD 中(左)和(右)菌群失调的持续时间和间隔分布的 Kaplan-Meier 曲线。两者都是近似指数的(以虚线拟合),UC 的平均值分别为 4.1 和 17.2 周,CD 的平均值分别为 7.8 和 12.8 周(参见 方法)。f, 与非生物样本相比,显着不同的宏基因组物种( 来自 130 名受试者的n = 1,595 个样本)、代谢物( 来自 106 名受试者的n = 546 个样本)和微生物转录物(来自 106 名受试者的n = 818 个样本)的相对丰度分布来自同一疾病组的菌群失调样本(Wald 检验;所有 FDR P < 0.05;补充表中的完整结果15 – 28 )。还显示了 ANCA、ASCA(IgG 或 IgA)、抗 OmpC 和抗 CBir1 抗体( 来自 61 名受试者的n = 146 个样本)的抗体滴度。箱线图显示中位数和下/上四分位数;胡须显示内部栅栏;盒子上方的样本大小。
值得注意的是,没有宏基因组物种是校正多重假设检验(补充表后从与IBD的个体的样品和来自对照个体之间不同显著1),与以前的工作相反4,5,18。我们假设这是由于研究参与者分为两个子集,一个具有相对不活跃的 IBD(由于缓解或最近发作),另一个具有更大的活动。这种分化已在多个 IBD 患者队列中观察到5 , 18,但这里更明显,因为我们没有专门从为活动性疾病选择的受试者中采集样本。因此,我们将具有与非 IBD 对照样品高度不同的分类组成的样品分类为“生态失调”(图2c,扩展数据图3a-e,参见 方法)。该队列中的菌群失调与疾病位置不符(例如,回肠 CD;F检验P = 0.11,参见 方法),并且在受试者体内纵向发生;它们与患者报告的疾病活动度和分子测量值弱相关(图2d,扩展数据图3a))。在 78 个完整的生态失调时期和 9 个审查期(即在时间序列结束时出现失调的受试者,参见方法),总共采集了 272 个失调样本 ,占所有样本的 17.1%(n = 178 (24.3 %) 在 CD 和n = 51 (11.6%) 在 UC)。失调时期的持续时间和时间图近似为指数,表明过渡至少部分是由随时间具有恒定概率的事件触发的(因此可能是随机的;图2e)。
使用所得到的生态失调的定义,dysbiotic周期对应于变化的在比没有整体IBD的表型(图的所有测量类型的较大部分1f中,补充表格15 - 28); 这可能反映出随着时间的推移,在极其异质的受试者中,活动性和活动性较低的疾病状态之间的划分更加清晰。尽管尚不清楚生态失调的哪些方面是 IBD 的原因或后果,但对这些变化的表征将有助于更好地了解疾病中的微生物动力学。与之前已确定疾病的横断面研究一样4,来自 CD 个体的失调和非失调样品之间的差异比来自 UC 个体的样品更明显(图1f)。值得注意的是,生态失调还区分了独立的宿主指标,例如具有高和低 ASCA(抗酿酒酵母抗体)、ANCA(抗中性粒细胞胞质抗体)、OmpC(外膜蛋白 C)和 CBir1(抗鞭毛蛋白)的个体血清学谱中的抗体滴度(图2f;Fisher 联合概率检验P = 0.00044,来自失调和非失调 CD 之间的 Wilcoxon 检验)。生态失调与人口统计学或药物治疗没有显着相关性(以受试者为随机效应的逻辑回归,所有 FDR P > 0.05)。生态失调概括了活动性疾病中已知的 α 多样性降低,但我们也将许多具有正常复杂性的群落确定为生态失调(扩展数据图4a)。值得注意的是,生态失调期间的分类学扰动反映了先前在 IBD 中横断面观察到的那些扰动6,例如CD 中包括Faecalibacterium prausnitzii和Roseburia hominis在内的专性厌氧菌的消耗以及大肠杆菌等兼性厌氧菌的富集(图2f,扩展数据)图4b )。瘤胃球菌属扭矩和瘤胃球菌属gnavus,在IBD两个突出的物种19, 分别在生态失调 CD 和 UC 中的丰度也不同(FDR P = 0.041 和 0.0087。一小部分物种的转录活性也显着增加(相对于基因组丰度的平均总转录本相对丰度;参见 方法)以及显示差异大量,包括梭状芽孢杆菌、梭状芽孢杆菌和R. gnavus(图2f)。在生态失调期间 ,它们的表达均显着增加(所有 FDR P < 0.07),因此它们在 IBD 中的作用可能比仅通过它们所暗示的更显着基因组丰度的差异。
在代谢组中,
- SCFAs 通常在生态失调中减少(图2f)。特别是丁酸盐的减少与先前观察到的丁酸盐生产者6如F. prausnitzii和R. hominis 的消耗一致,这也在此处观察到(图2f)。
- 我们还检测到CD 参与者的生态系统失调样本中的初级胆汁酸及其甘氨酸和牛磺酸结合物(甘氨胆酸q = 5.2 × 10 -5,牛磺胆酸q = 1.3 × 10 -5)的富集,与非失调样本相比.
- 类似地,甘草脱氧胆酸盐 ( q = 1.1 × 10 -4) 也得到了丰富。
- 相比之下,次级胆汁酸石胆酸盐和脱氧胆酸盐(分别为q = 5 × 10 -7和q = 1.8 × 10 -4)在生态失调中减少,表明次级胆汁酸产生菌在 IBD 相关的生态失调中耗尽,或者通过结肠的传输时间太短,这些化合物不能被代谢20 , 21。
微生物失调期间这些显着的代谢组学差异与疾病期间预期的变化一致,提供了进一步的证据,证明失调措施与 IBD 尤其相关。
我们还在生态失调期间观察到了几个以前未描述的生化差异,例如酰基肉碱水平的巨大变化。
- 许多酰基肉碱在生态失调中显着富集(所有 FDR P < 0.05;参见扩展数据图4c),而基础代谢物的水平通常降低(图2f,扩展数据图4d)。然而,值得注意的是,花生四烯酰肉碱(C20:4 肉碱)减少,游离花生四烯酸(参与炎症的前列腺素前体)增加(图2a)。
- 像胆汁酸,肉碱是微生物改性,可以有竞争取决于确切的修饰的表型作用的化合物:升例如,肉碱往往具有抗炎作用,而结合脂肪酸的肉碱对肠道炎症的作用并不统一22。这些生化相关代谢物的相反变化进一步表明,在生态失调期间观察到的差异不仅仅源于粪便的大量稀释。许多其他代谢物在患有失调型 IBD 的个体中也发生了显着改变(548 种已知代谢物中的 117 种,FDR P < 0.05;扩展数据图4d,补充表16),表明代谢物池与宿主和微生物组特定的分类和分子特征(图2f)。最后,虽然我们发现只有一个单一的、特征不佳的噬菌体在 IBD 和生态失调中普遍存在差异(特别是 IBD 的患病率降低;补充表3、17),但我们注意到一些参与者在出现生态失调之前表现出病毒载量峰值期间(补充图2)。
三、IBD 肠道微生物组稳定性降低
我们针对来自许多受试者的粪便衍生多组学的密集时间序列使我们能够进行深入的纵向分析,整合微生物组的多项测量。随着时间的推移,每个受试者的微生物组倾向于从宏基因组、宏转录组和代谢组学特征的基线偏离更多(图3a;F检验幂律拟合P < 10 -24;参见 方法)。这些变化对于患有 CD 和 UC 的个体的分类学特征最为明显(幂律拟合的F检验差异P < 10 -9),其中一个个体的微生物组在较早的时间点可能几乎没有与自身相同的物种(差异为 1;图3a),与之前的观察结果一致9。物种内总结的转录本(扩展数据图5a)显示出与宏基因组物种丰度相似的趋势(所有F检验P < 8 × 10 -4)。同时,基因家族转录本(Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthologues (KOs))、代谢物(图3a)和蛋白质(KOs,扩展数据图5a)) 的变化要快得多,大约两周后的变化基本上与较长时间段内的变化一样多(增加趋势较小或不显着:非 IBD、UC 和 CD F检验P = 0.0006、0.001 和 0.04,分别用于转录本;代谢物为 0.02、0.06 和 0.003;蛋白质组学为 0.5、0.15 和 0.06)。这表明这些特征在患有和不患有 IBD 的个体的肠道中变化很快,并且在疾病期间缺乏额外的、更极端的偏移。
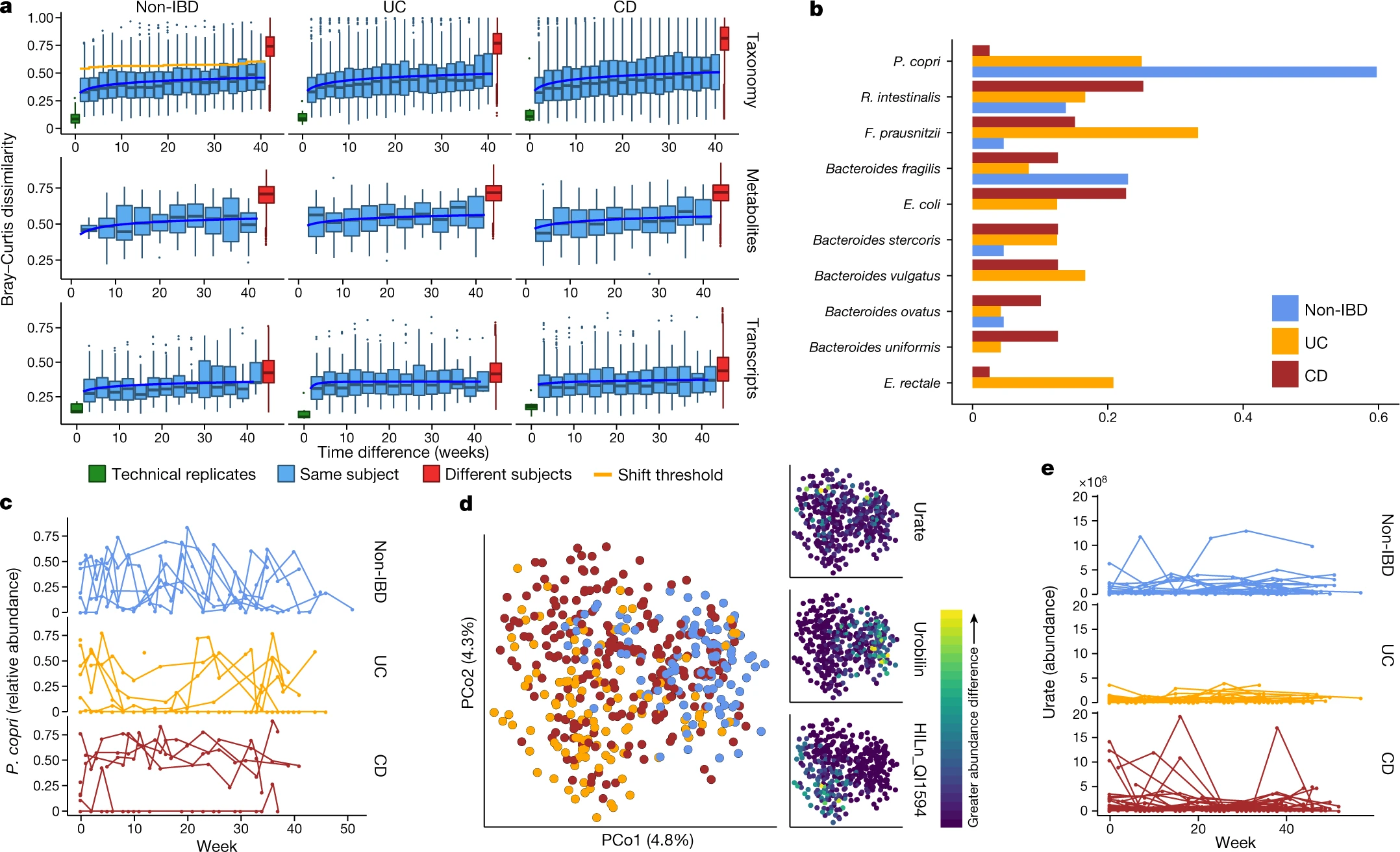
a,与不同的人或技术复制相比,受试者内的布雷-柯蒂斯差异作为干预时间差异的函数;计算宏基因组分类谱(物种;n = 130 名受试者的 1,595 个样本)、代谢组学(n = 106 名受试者的 546 个样本)和功能谱(KO 30 个基因家族;n = 106 名受试者的 818 个样本)。箱线图显示中位数和下/上四分位数;胡须显示内部栅栏。蓝色,最小二乘幂律拟合;橙色,微生物组变化的阈值(参见 方法)。扩展数据中的蛋白质组学和物种水平转录本 图5a. UC 和 CD 的受试者内变化比非 IBD 的分类学特征(F检验P 分别 为 3.9 × 10 -10和 1.2 × 10 -18)和转录本(P = 0.00016 和 1.7 × 10 -5),代谢物的混合差异(P = 0.012 和 0.23)。在 0 周时显示技术重复(如果可能)。b,在轮班期间变化最大的前 10 种物种的轮班频率,按轮班次数排序为主要贡献者,按疾病表型分层(完整表格补充表29)。c , P. copri对关节炎23和国际人群感兴趣44,它单独在 CD 中保持稳定的丰度,但在对照中保持大量松弛动力学(连续时间点之间的绝对差异的双尾威尔科克森检验P = 4.2 × 10 -6非 IBD 之间和 UC,以及非 IBD 和 CD 之间的1.1 × 10 -4)。绘图显示 22 名受试者至少有一个时间点的差异丰度超过 10%(n = 267 个样本)。d,基于代谢组学(标准化绝对丰度差异的 Bray-Curtis 主坐标)对个体内时间相邻样本的排序。疾病组显着分离 ( n = 来自 106 个受试者的 440 个样本对;PERMANOVA R 2 = 2.8%,P < 10 -4 )。尿胆素、尿酸盐和未识别的非靶向特征与 PCoA 中的疾病组分离(右);HILn_QI1594(HILIC-neg 方法m / z = 152.0354,RT = 4.16 分钟)。e,与c 相同,但对于尿酸盐(双尾 Wilcoxon 检验P = 0.0012 非 IBD-UC,P = 0.044 非 IBD-CD;n = 106 名受试者的 546 个样本)。
我们通过搜索连续时间点之间微生物组的“变化”进一步表征了大规模时间差异,定义为 Bray-Curtis 差异与不同人之间的差异比一个人内部的差异更相似(图3a,扩展数据图5b,见 方法)。首先,仅考虑宏基因组分类学概况,我们发现了 166 个这样的变化,其中 39 个在没有 IBD 的个体中(总共 382 个可能),44 个在 UC 个体中(381 个),83 个在 CD 个体中(650 个)(补充表29)。由于总观察时间的差异,CD 或 UC 患者的轮班率仅略高于非 IBD 参与者(分别为每年 2.09 和 1.83 轮,而每年轮班为 1.79),并且这些通常仅限于失调个体的亚群(图3a)。然而,相对丰度变化最大的物种差异显着(图3b)。没有 IBD 的个体的变化主要发生在具有高丰度普氏菌的个体中,这些个体在数周到数月的过程中经历了反复的扩张和松弛循环(图3c))。这种有机体特别令人感兴趣,因为它的行为是人口规模的外群,并且在新发类风湿性关节炎23期间富集。IBD 参与者中缺乏由P. copri引起的变化并不是由于这些个体中不存在P. copri或没有 IBD 的个体过多(27 名非 IBD 受试者中有 6 名至少有一个时间点超过10% P. copri,与健康人群一致10 , 24 )。相反,IBD 人群中存在的相对丰度保持更稳定(图3c)。IBD 参与者的分类学变化反映了早期对专性厌氧菌相对减少和兼性厌氧菌过度生长的观察结果(图3b,扩展数据图5c),并且经常与进入和退出生态失调相对应(28 和 23 轮班标志着进入和分别在 IBD 中退出,占 IBD 转移的 40%)。大肠杆菌尤其对 IBD 的大量变化做出了贡献,尽管没有明确的模式与哪些物种进行丰度交易(扩展数据图5c、d)。
当我们以类似的方式定义代谢组学谱的变化时(扩展数据图5e),变化率大约是宏基因组的一半(没有 IBD 的参与者每年 1.05 次转变,UC 中每年 0.99 次转变和 1.36 次转变每年在 CD 中),尽管这些数据受到较少代谢组学样本可用性的强烈影响(扩展数据图5e)。我们检查了来自同一受试者的相邻样品之间代谢物谱的差异,发现诊断有显着的分离(图3d;PERMANOVA P < 10 -4)。这些差异主要是由未知化合物驱动的,强调需要进一步的化合物注释工作和后续工作以确定这些化合物在 IBD 中的重要性。差异最大的特征包括尿胆素(在没有 IBD 的个体中表现出更大的差异)、尿酸盐(主要在 CD 患者中),以及m / z为 152.0354 和保留时间 (RT) 为 4.16 分钟的特征(可能是甲酸吡啶醛的酸加合物),这解释了主要针对 UC 的差异。转变的主要贡献者主要是未识别的化合物(扩展数据图5f,补充表30)。HILp_QI22918,m / z未知特征648.43067 和 5.03 分钟的 RT,仅在 IBD 患者中贡献最多(十个)变化。在已知化合物中,甲基咪唑乙酸和尿酸盐是造成最大位移的主要因素(各有四个位移;图3e)。
四、微生物组相关宿主因素
当我们将宿主分子测量值(主要来自基线时结肠镜检查的肠道活检)纳入我们对 IBD 微生物组的分析中时,对种群变异性的主要影响与单独影响微生物群的那些显着不同。特别是,即使面对微生物变异25(扩展数据图2d),组织定位也是肠上皮基因表达的主要驱动因素(扩展数据图2c)。因此,我们对每个标准化活检位置独立进行了微生物组和表型关联分析(参见 方法)。
与没有 IBD 的个体相比,我们在回肠(来自 CD 患者)和直肠(CD 和 UC)发炎部位的患者活检组织中鉴定了显着差异表达(DEG)的基因(扩展数据图6a)。该分析确定了 305 和 920 个基因,分别在回肠和直肠中差异表达(主要是过表达)的基因,用于进一步分析(共代表 1,008 个独特基因,负二项式模型 FDR P < 0.05 和倍数变化 >1.5;图。图4a,补充表31)。这些包括可以直接影响共生微生物的基因,例如抗菌CXCL6(一种细胞膜破坏因子26) 和SAA2(抑制革兰氏阴性菌的生长27),以及间接微生物调节剂,如DUOX2(产生活性氧28)和LCN2(通过螯合诱导微生物铁饥饿29;图4b)。对 DEG中 KEGG 30通路过度表达的富集分析测试也证实了免疫相关通路的强代表(单侧超几何测试,FDR P < 0.05)。尤其是 IL-17 信号通路,其成分先前已在 CD 患者回肠活检的基因表达研究中确定31,32,是在两个回肠和直肠上调DEGS富集的(FDR P = 2.8×10 -14 ;图4a中,补充表32)。在 UC 患者的直肠活检中上调的 DEG 中,我们发现补体级联进一步富集(FDR P = 4.4 × 10 -10),这是先天免疫的一个组成部分33,与 IBD 有关联25 , 34 , 35。
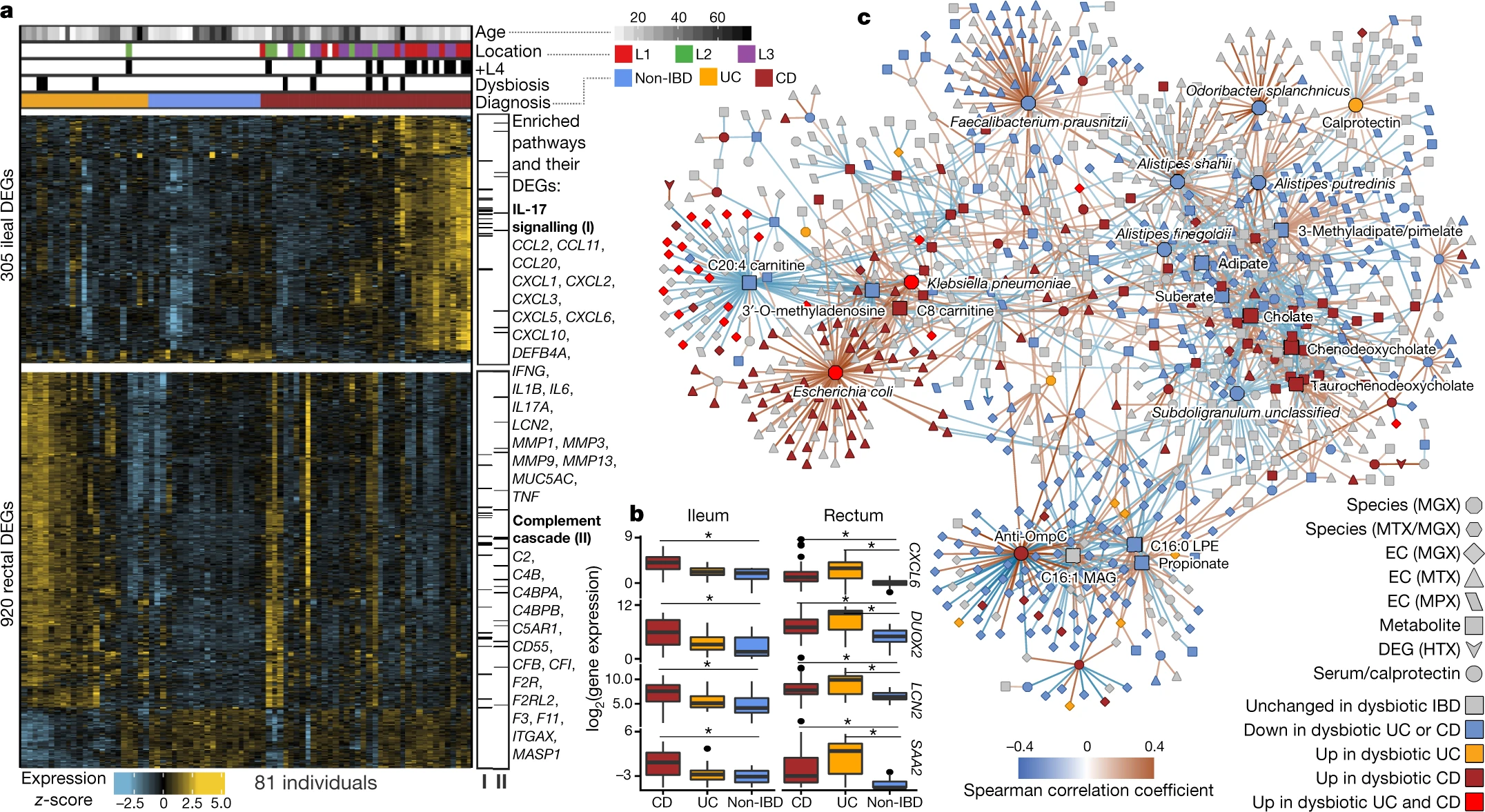 为了确定与这些变化最相关的微生物组成分,我们测试了与使用 16S 扩增子测序直接从相同标本测量的微生物相对丰度共变的转录本。我们分别在回肠和直肠中确定了 31 个和 106 个显着的基因-操作分类单元 (OTU) 对,两个位点之间没有重叠,这与将它们分开的不同整体基因表达模式一致(部分 Spearman 相关 FDR P < 0.05 ;参见 方法,扩展数据图6b,补充表33)。涉及的基因包括已知的 IBD 相关宿主-微生物相互作用因子,包括DUOX2及其成熟因子DUOXA2 31 , 36,这两者都与回肠中瘤胃球菌科 UCG 005 (OTU 89) 的丰度呈负相关。几个趋化因子基因的表达,其中一些已经报道了抗菌特性37(CXCL6,CCL20),与回肠中直肠真杆菌(OTU 120) 和链球菌(OTU 37) 和Eikenella (OTU 39 )的相对丰度呈负相关) 在直肠中,表明这些物种对这些趋化因子的活性最敏感。最后,虽然这个队列不是为基因关联发现而设计的(补充讨论,扩展数据图6c,d,补充表34),我们还提供了92名受试者的外显子组测序,未来可能会与更大的人群整合。
为了确定与这些变化最相关的微生物组成分,我们测试了与使用 16S 扩增子测序直接从相同标本测量的微生物相对丰度共变的转录本。我们分别在回肠和直肠中确定了 31 个和 106 个显着的基因-操作分类单元 (OTU) 对,两个位点之间没有重叠,这与将它们分开的不同整体基因表达模式一致(部分 Spearman 相关 FDR P < 0.05 ;参见 方法,扩展数据图6b,补充表33)。涉及的基因包括已知的 IBD 相关宿主-微生物相互作用因子,包括DUOX2及其成熟因子DUOXA2 31 , 36,这两者都与回肠中瘤胃球菌科 UCG 005 (OTU 89) 的丰度呈负相关。几个趋化因子基因的表达,其中一些已经报道了抗菌特性37(CXCL6,CCL20),与回肠中直肠真杆菌(OTU 120) 和链球菌(OTU 37) 和Eikenella (OTU 39 )的相对丰度呈负相关) 在直肠中,表明这些物种对这些趋化因子的活性最敏感。最后,虽然这个队列不是为基因关联发现而设计的(补充讨论,扩展数据图6c,d,补充表34),我们还提供了92名受试者的外显子组测序,未来可能会与更大的人群整合。
五、动态的、多组学微生物组相互作用
我们接下来通过构建一个大规模交叉测量类型关联网络来搜索可能是 IBD 疾病活动基础的宿主和微生物分子相互作用,该网络结合了十种微生物组测量:宏基因组物种、物种水平转录率、作为酶委员会捕获的功能谱。 EC) 基因家族(MGX、MTX 和 MPX)、代谢物、宿主转录(分别为直肠和回肠)、血清学和粪便钙卫蛋白。为了确定微生物组成分之间的共变,超出与炎症和疾病状态严格相关的成分,首先使用相同的混合效应模型(或适当时的线性模型)对每种测量类型进行残差,用于确定差异丰度(“调整' 网络;见 方法)。这种残差使用纵向测量来最小化任何个体间的差异(包括 IBD 状态),以及作为检测到的关联的驱动因素的生态失调偏移,从而随着时间的推移突出人内关联。由此产生的网络包含 53,161 条重要边(FDR P < 0.05)和 2,916 个节点,跨越所有测量类型的特征(补充表35)。我们构建了一个过滤子网络,用于从每种测量类型的前 300 个边缘(按P值)进行可视化,其中至少一个连接节点与生态失调相关(图4c)。
来自五个粪便衍生测量的代表作为该网络中的枢纽(定义为具有至少 20 个连接的节点),所有这些都被确定为在生态失调中差异丰富。特别相关的分类特征(来自宏基因组和宏转录组)包括F. prausnitzii的丰度和与Subdoligranulum 38相关的未分类进化枝的丰度,它们在系统发育上密切相关,尽管两种生物体共有的唯一分子特征与胆固醇和肌苷的丰度共变(扩展数据图7a )。F. prausnitzii占总体上一些最强的关联,包括在生态失调中下调的许多 EC 的表达。另一方面,大肠杆菌(以及较小程度的副流感嗜血杆菌)占上调 EC 的很大一部分。Roseburia属的成员在宏转录和宏基因组学上也与胆汁酸和许多酰基肉碱相关,这表明Roseburia(与Subdoligranulum一起)参与了 IBD 中观察到的肉碱和胆汁酸失调。
酰基肉碱和胆汁酸作为整体化学类别在网络中占有突出地位,部分与它们在生态失调期间的变化有关。酰基肉碱与许多与生态失调相关的物种有关,包括R. hominis(九个酰基肉碱,FDR P < 0.05;补充表35)、肺炎克雷伯菌(三个)和副流感嗜血杆菌(三个),以及C.bolteae 的表达(三),表明涉及多个调节尺度,包括基于长期生长和短期转录。网络中特别值得注意的生化中心包括 C8 肉碱,另一种酰基肉碱在失调的 CD、胆酸盐、鹅去氧胆酸盐和牛磺鹅去氧胆酸盐中显着增加,它们一起占 107 个边缘(6%;图4c))。其他突出的代谢物关联包括几个长链脂质中心和 SCFA 丙酸盐;针对 OmpC 的抗体与这些以及参与系统生物合成或作为相互作用物的众多 EC 的宏基因组丰度密切相关。钙卫蛋白作为其自身测量类型中的唯一特征,与一些在生态失调中没有差异的代谢物以及一些与生态失调相关的 ECs 的宏基因组丰度微弱相关。在这个高显着性子网络中出现了三个宿主基因:GIP、NXPE4和ANXA10 的回肠表达. RNA聚合酶的表达也是网络中的一个突出节点,虽然不是枢纽,但在生态失调中上调(扩展数据图8)。这种必需酶类的调节依赖于生长速率39,这表明微生物群落作为一个整体在失调的 IBD 中更常处于较高的生长条件下。
最后,我们还确定了考虑到生态失调的微生物组特征之间的关联,从而产生了使用相同方法但未针对生态失调进行调整的第二个网络(“未调整”;补充讨论,扩展数据图7b、9,补充表36)。总之,这些网络将在 IBD 中观察到的多种微生物组破坏与许多分子特征类型之间的关联联系起来,这些分子特征类型代表了对 IBD 和胃肠道炎症的潜在机制进行后续研究的潜在目标。
六、结论
作为 HMP2 的一部分,我们开发了 IBDMDB,这是对与 IBD 动力学有关的肠道微生物组的多个分子特征的首批综合研究之一。虽然在微生物组测量(宏基因组学、宏转录组学、代谢组学等)中,总体人口结构具有可比性,但每次测量都确定了 CD 和 UC 中纵向生态失调的互补分子成分。之前的研究已经捕捉到了一些,例如有利于耐氧、促炎进化枝的分类学变化;其他的,例如疾病期间梭状芽孢杆菌的更多基因表达,是通过使用新的测量(宏转录组)发现的。多个微生物组测量的时间稳定性同样因 IBD 表型和疾病活动而异,P. copri在 IBD患者中)。除了数据、协议和相关生物信息学方法之外,我们的数据还提供了在 IBD 期间被确定为潜在核心的多组学特征之间的新关系目录,以支持未来的研究。
通过利用微生物组的多组学视图,我们的结果挑选出许多宿主和微生物特征进行后续表征。一个未分类的Subdoligranulum物种,最近显示形成一个新物种级进化枝的复合体38,在 IBD 中显着减少,并且是功能网络的核心,与广泛的 IBD 相关代谢物相关联,两者都可识别(例如,胆汁酸和多不饱和脂肪酸)且无法识别。该进化枝可能包含至少七个与Subdoligranulum、Gemmiger和Faecalibacterium属密切相关的物种,通常被认为是有益的丁酸盐生产者,特别是在 IBD 中40 . 因此,额外物种的分离和表征——尤其是与这些相关代谢物的结合——可能会揭示这些进化枝的生理和免疫相互作用以及它们在 IBD 中消耗的后果。更一般地说,有关微生物的菌株水平分析仍有待进行,特别是与宿主上皮细胞和相应的分子变化直接相关的情况。这种分析对于本研究的现有数据是可行的,并将有助于查明导致 IBD 相关初级未结合胆汁酸积累和次级胆汁酸消耗的特定生物体41。目前已知只有极少数、低丰度的物种能够进行次级胆汁酸代谢42,并扩大已知携带适当代谢盒的菌株范围将表明治疗恢复的潜在新目标。除了短链脂肪酸和胆汁酸,这里观察到的大规模酰基肉碱生态失调也可能为 IBD 提供一个有希望的新靶点,特别是在确定代谢物库的这种转变是宿主驱动还是微生物组驱动之后。
我们强调,尚未确定微生物组的这些多组学特征是否可以在疾病发生之前预测疾病事件,并且尚未确定不同分子事件的疾病相关时间尺度(例如,静态宿主遗传学,相对缓慢的表观遗传学或微生物生长,快速的宿主和微生物转录变化)。寻找与特定受试者基线状态的最早偏离也可能是富有成效的,虽然它们本身仍然是“有益的”,但可以预测随后出现的生态失调或疾病症状。在本研究的数据中可能有一些这样的特征,尽管在更细粒度的时间尺度或使用干预性研究设计可能更好地进行其他因果分析。最重要的是将这些分子结果带回临床,以更好地预测 IBD 进展和结果的生物标志物的形式,并作为一组新的宿主 - 微生物相互作用目标,可以开发用于改善疾病的治疗方法。
七、方法
参考资料
- Nature volume 569, pages655–662(2019). Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. https://www.nature.com/articles/s41586-019-1237-9
