【3.7.4】蛋白质蛋白质界面的hotspot,走向药物发现
鉴定改变蛋白质蛋白质相互作用的药物样小分子可能是药物发现的关键步骤。 然而,找到靶向蛋白质复合物中的界面区域的这种分子是非常具有挑战性的。 最近的研究结果表明,这种分子通常在蛋白质蛋白质界面中特异性地靶向能量上有利的残基(hot spots)。 这些残基有助于蛋白质蛋白质复合物的稳定性。 绑定和未绑定结构上hot spots的计算预测可能对于在目标界面上找到可药物位点是有用的。 我们在该综述
- 第一部分回顾了计算热点预测方法的最新进展
- 然后提供了热点在药物设计中如何至关重要的实例。
一、前言
蛋白质蛋白质相互作用在调节生物过程,细胞和信号传导途径中起着至关重要的作用。 蛋白质蛋白结合位点称为界面(interfaces)。 界面中的残基特性是蛋白质蛋白质识别,结合和亲和力的关键要素。 基于残基的分析可以帮助揭示蛋白质蛋白质结合机制。 天然蛋白质蛋白质相互作用的改变可能导致几种疾病。 因此,靶向蛋白质之间的界面在药物发现中具有巨大的潜力(Kar等人,2012; Thangudu等人,2012; Wells和McClendon,2007)。 靶向蛋白质蛋白质相互作用的药物如果不是变构位点,最终应该与蛋白质界面结合。 然而,靶向界面比在药物发现中靶向酶或G蛋白偶联受体的活性位点更具挑战性,因为界面相对较大,通常是扁平的而没有特异性配体结合口袋。
蛋白质蛋白质界面中的残基不能同等地促成结合能。有一些称为热点(hot splots)的关键残基对结合能的贡献最大(Bogan和Thorn,1998; Clackson和Wells,1995)。可以通过丙氨酸扫描诱变实验检测热点残基(Clackson和Wells,1995)。如果在将残基突变为丙氨酸后结合能差大于2kcal / mol,则将其标记为热点。 Bogan和Thorn(Bogan和Thorn,1998)分析了热点的氨基酸组成,并得出结论,一些残基更有利于蛋白质蛋白质界面中的热点。 Tyr,Arg和Trp是最常见的,由于它们的大小和构象而至关重要。他们还报告说,热点被一组残基包围,这些残基在能量上不太重要,类似于管道配件中的O形环,以阻塞水分子中的热点。可及表面积的变化与残基的能量贡献之间存在相关性(Guharoy和Chakrabarti,2005)。 Moreira等人(2007a)也支持使用分子动力学(MD)模拟的O形环假设。此外,这些热点不是随机分布在界面中而是聚集在一起。热点在密集区域内组装。这些模块化装配区域称为热区(hot regions)(Keskin等,2005)。该结合位点组织证明给定蛋白质分子如何与不同蛋白质配偶体结合。 Kleanthous及其同事(Meenan等,2010)表明,大肠杆菌E9核酸内切酶和免疫蛋白9的同源复合物界面上的有限数量的突变提供了E9与免疫蛋白2的高亲和力结合,尽管比较弱的亲和力同源复杂。这些实验结果也由计算热点组织研究(Cukuroglu等,2012)。热点残基的组织提供了获得对不同伴侣的结合亲和力和特异性的机制。因此,这些残基的协同性可以揭示特异性的复杂结合组织(Cukuroglu等人,2010; Shulman-Peleg等人,2007)。
热点对找到蛋白质蛋白质复合物的动力学行为也很重要。 Agius等(2013)利用热点能量学和热点/热区的结构来预测突变时解离速率的变化。 他们使用一组生物物理和统计描述符来估算热点能量。 然后将这些描述符用作评估单个或多点突变的成功解除率变化的特征。
通过实验技术确定热点残基是昂贵且耗时的,因此已经开发了计算方法来预测结合和未结合蛋白质结构中的热点残基。 预测结果表明,多年来预测精度都在提高。 预测方法将是对实验研究的补充,以发现对结合亲和力和特异性具有重要作用的热点残基。 此外,计算方法可以指导实验突变研究,并解释蛋白质结合的功能和机制方面(Fernandez-Recio,2011)。
最后但并非最不重要的是,越来越多的研究表明热点在药物设计中可能很重要(Fry,2012; Jubb等,2012; Thangudu等,2012; Wells和McClendon,2007)。 药物样小分子更喜欢与蛋白质蛋白质界面的热点结合(Arkin和Wells,2004)。 下面,我们将首先回顾计算热点预测和蛋白质 - 药物相互作用的最新进展以及热点在药物设计中的关键性。
二、预测热点残基
热点预测方法可分为两类:
- 基于未结合的单体结构
- 蛋白质蛋白复合物结构。
以前的研究使用不同的数据集和阈值来预测热点残基。 可用工具及其属性的完整列表在SupplementaryFile_1中列出。
三、结合蛋白质结构的热点预测
大多数热点预测工具集中于结合的蛋白质蛋白质相互作用,以检测蛋白质界面中的热点残基。通过实验技术检测的热点残基的分析仅限于少量复合物。关于实验确定的热点残基的信息已经存储在数据库中。 ASEdb是Thorn和Bogan(2001)开发的第一个丙氨酸突变数据库,后来BID由Fischer(2003年)等人开发。通过实验确定热点残基的成本和难度,导致通过计算方法预测这些残基。因此,Kortemme和Baker(Kortemme和Baker,2002; Kortemme等,2004)提出了一种计算丙氨酸扫描方法,该方法使用填充相互作用,氢键和溶剂化的能量。 Guerois等。 (2002)使用FOLD-X能量来预测热点残基。另一种基于能量的方法由Gao等人(2004)使用氢键,疏水和VdW相互作用(三种主要的非共价相互作用)开发,以估计每个界面残基对结合能的个体贡献。计算出的突变能量变化与实验结果相符。
MD模拟适用于原子水平上蛋白质蛋白质相互作用的详细分析,它们可用于预测热点残基(Gonzalez-Ruiz和Gohlke,2006; Grosdidier和Fernandez-Recio,2008; Huo等,2002; Landon 等人,2007; Moreira等人,2007a; Rajamani等人,2004; Wang等人,2013; Yogurtcu等人,2008)。 能量和基于MD的热点预测方法都具有高准确率,但它们在计算上很昂贵并且难以应用于大规模研究。
基于知识的方法形成了另一种预测热点残基的方法。他们通常使用机器学习方法来学习已知的热点数据。基于知识的方法的主要优点是它们的计算效率。然而,它们对残基类型,尺寸,疏水性,可接触表面积等特征的选择非常敏感,以表征热点残基,并且很难找到最佳特征组合。大多数研究使用不同的特征来提高它们的预测精度,即使它们使用类似的机器学习算法,例如支持向量机(SVM),线性回归,神经网络,贝叶斯网络和随机森林模型。Assi等人(2010)使用序列保守,能量分数和contact number信息来预测热点残基。 Lise等人建立了一种基于机器学习的预测方法(Lise et al,2009),但他们的方法在Arg和Glu上效果不佳,因此他们改进了他们的方法,增加了两种特定于这两种氨基酸的分类器(Lise et al 2011) 。他们主要使用范德华力,去溶剂化,氢键和静电能量来预测热点残基。 Koes和Camacho(Koes和Camacho,2012)使用机器学习方法,可获得的表面积(accessible surface area,ASA),相对ASA(RASA),进化速率,保守评分,络合的自由能和丙氨酸突变值的自由能变化。他们报告说ASA,RASA和每个残基估算的自由能值是最具信息量的特征,并具有良好的分类准确性。此外,Xia等人(2010)详尽地研究了蛋白质结构的不同特征以提高热点预测的准确性,并且他们得出结论,ASA相关特征表现出更好的判别力,如Cho(2009年)等人所建议的。 根据Xia(2010)等人的工作,突出指数也是一个很好的热点鉴别器,这也在Li等人的着作中得到了证明(Li et al。,2004)。与这些特征不同,Cho等人(2009)发现加权原子堆积密度和加权疏水性对热点残基预测具有判别力。 Mitchell和她的同事提出了两种不同的方法:KFC(Darnell等,2007),它使用界面残基的形状特异性和生化接触特征,并用KFC2(Zhu和Mitchell,2011)更新了它们的方法,使用了界面溶剂化,原子密度和可塑性特征。他们的结论是,缺乏可塑性强烈表明存在热点残基,但这不是必需的。王等人(2012a)使用残基的质量,极化率和等电点,相对侧链可及表面积和平均深度指数来预测热点残基。如先前的研究所示,使用可接触的表面积,能量,原子堆积密度和塑性相关特征以合适的组合提高了热点预测精度。
还使用基于经验公式的方法代替机器学习算法来预测热点残基。 Pavelka等(2009)仅使用保护分数来识别热点残基。 Guney等人(2008)使用保守分数和ASA值与经验公式。 Tuncbag等(2009)表明,RASA和成对潜力比预测的保守分数更具辨别力。实际上,应该是蛋白质家族决定保护是否具有歧视性。对于抗体抗原复合物,保存(conservation)不是一个好的特征(Assi等,2010)。 Kruger和Gohlke(2010)使用具有埋藏度的成对电位来预测热点残基。 Geppert等(2011)使用成对电势,原子类型和残基特性来生成具有额外投票系统的经验公式,以便找到功能性热点残基。 Shulman-Peleg等人(2007)进行功能相似的蛋白质蛋白质复合物的结构比对,以找到对应于热点残基的残基的空间化学保守性。蛋白质蛋白质界面的热区域在功能上很重要,之前的研究使用序列和结构信息来找到这些区域(Armon等,2001; Cukuroglu等,2012; Hsu等,2007)
为了预测热点残基,网络拓扑属性变得突出。 Del Sol和O’Meara(del Sol和O’Meara,2005)将网络拓扑结构应用于蛋白质复合物的残基网络,以找到热点残基。 他们指出网络中高度中心的残基很可能是热点残基。 Li和Liu(2009)从复合物的两个单体生成了一个二分图,并找到了来自最大biclique子图的双重图案,以识别热点残基。 Tuncbag等(2010b)解释了残基对minimum cut trees中几个mincuts的贡献,以定义热点残基。
四、未结合蛋白质结构的热点预测
有三项基于未结合蛋白质结构的研究。 ISIS(Ofran和Rost,2007)是第一个使用氨基酸序列预测未结合蛋白质结构上的热点残基的预测工具。 pyDockNIP(Grosdidier和Fernandez-Recio,2008)使用归一化界面倾向值,该值来自刚体对接与静电和去溶剂化评分,用于预测相互作用热点。在这项工作中,他们使用复杂形式的蛋白质来推导其预测功能;但是,此功能可用于查找未绑定形式的热点残基。 Ozbek等人(2013)使用从高斯网络模型(GNM)获得的高频模式中的动态波动来预测未结合结构上的热点残留。如果存在未结合形式的结构GNM(Haliloglu和Erman,2009; Ozbek等,2013)表现更好,但如果存在唯一的序列信息,ISIS(Ofran和Rost,2007)具有优势。关于未结合结构上残基的热点预测的研究数量有限。需要做很多工作来预测蛋白质界面上的热点,具有高精度和特异性
五、多界面结合和热点残基
一些蛋白质可以在其表面上使用相同或不同的界面与多个伙伴相互作用。这些蛋白质不可能与使用不同界面的许多伙伴相互作用,因为它们具有有限的结合表面积。因此,他们反复使用一些结合位点(Keskin和Nussinov,2007; Kim等,2006)。 Thangudu等人(2012)表明这个多重结合位点至少有一个热点残基,这些位点通常重叠。多种相互作用的亲和力和特异性可源于不同热点残基的协同作用(Cukuroglu等,2012)。一项实验研究还强调,中枢蛋白SMAD3的不同热点残基的突变改变了与其配偶体相互作用的亲和力(Schiro等,2011)。热点残基分析可以揭示具有多界面结合的蛋白质的复杂结合策略。 Cukuroglu等人(2014)在PDB中用多界面结合策略提取所有可能的蛋白质(Berman等,2000)。这些结构都来自实验性的非冗余结构。根据分析结果,肌动蛋白使用40种不同的结合模式,同时与另一种肌动蛋白单体相互作用,如图1所示(叠加40种不同的复合物)。图1中心的蓝色单体是共同的肌动蛋白单体,而用不同颜色表示的其他肌动蛋白单体是相互作用的配偶体。当分析复合物的热点残基(热区域发现热点和热区域时(Cukuroglu等人,2012)),只有两个界面没有任何计算热点。 35个界面使用肌动蛋白表面上的67个不同的热点残基(图1中的蓝色),并且在产生结合亲和力和特异性的任何相互作用中没有重复热点残基组合。在三种复合物中,肌动蛋白单体(图1中的蓝色)即使其伴侣单体具有热点残基也没有任何热点残基(补充表1)。泛素使用22种不同的结合策略,同时与另一种遍在蛋白单体相互作用,如图2所示(叠加22种不同的复合物)。计算热点列于补充表2中,具有计算热点的复合物在图3中单独显示。泛素单体(图2中的蓝色)使用17种不同的残基作为热点,并使用这些热点残基在13个复合体中。在四种配合物中,泛素单体(图2中的蓝色)即使其伴侣单体具有热点残基也没有任何热点残基。只有五个复合物没有任何计算热点,其他17个具有热点残基的复合物如图3所示。图3的着色与图2相同。蓝色泛素单体使用不同的热点与其配偶体的残基组合以提供结合亲和力和特异性。对于这两个例子,界面中的热点残基主要形成为热区。对于肌动蛋白复合物,32种复合物中的27种复合物中的热点残基在靶标上具有热点残基并且伴侣蛋白质彼此接触,在遍在蛋白实例中,该情况发生在11种复合物中的10种复合物中。这些互补的热点相互作用将成为药物研究的有希望的目标。然而,即使热点在伴侣蛋白中没有任何互补的热点残基,它们也将是抑制蛋白质蛋白质相互作用的有希望的靶标。

图1 肌动蛋白单体以蓝色卡通表示。 其他颜色是肌动蛋白单体的相互作用配偶体(肌动蛋白和肌动蛋白相互作用)。 球和棒表示显示界面残基和大红球表示至少在肌动蛋白单体(如蓝色卡通表示中所示)的相互作用之一中用作热点的残基。 (VMD(Humphrey等人,1996)用于制备图)。
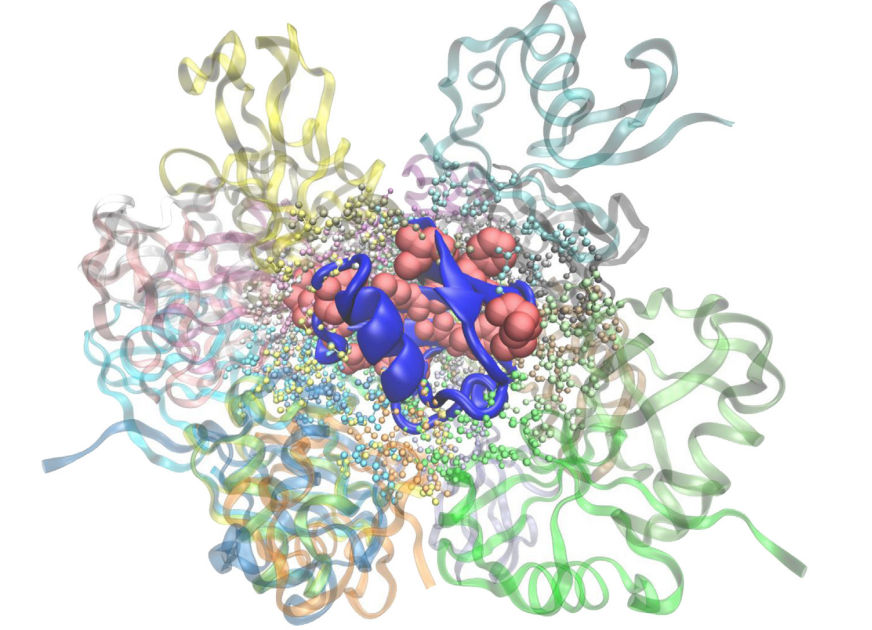
图2 遍在(Ubiquitin)蛋白单体以蓝色卡通表示。 其他颜色是泛素单体(泛素泛素相互作用)的相互作用伙伴。 球和棒表示显示界面残基和大红球表示显示至少在泛素单体(如蓝色卡通表示中所示)的相互作用之一中用作热点的残基。 (VMD(Humphrey等,1996)用于制备图)。
图3.遍在蛋白泛素(Ubiquitine Ubiquitin)与热点残基的相互作用分开表示。 热点残留物显示在补充表2中。卡通表示的颜色与图2相同。(VMD(Humphrey等,1996)用于制备图)。
六、蛋白质药物相互作用和热点
直到最近,药物基因组被认为仅由有限数量的蛋白质家族组成,如G蛋白偶联受体(GPCR),丝氨酸/苏氨酸和酪氨酸蛋白激酶,锌金属肽酶,丝氨酸蛋白酶,核激素受体和磷酸二酯酶。 这些药物靶组主要是酶,离子通道或受体,具有明确的和预先形成的凹形结合位点(Jubb,2012)。 然而,同一蛋白质家族成员之间的结构和序列相似性增加了药物滥交的可能性(Engin等,2014)。 此外,研究人员意识到,旨在生产高亲和力药物的基于靶标的药物设计方法产生低选择性(Jubb等,2012)。 人们越来越意识到药物滥交导致的损害(副作用)导致人们在药物发现中寻找新的策略。
自从发现热点在稳定蛋白质蛋白相互作用(proteineprotein interactions,PPIs)中的重要作用以来,已经广泛研究了针对热点的小分子对PPI的抑制作用。由于生物功能是通过蛋白质蛋白质相互作用(PPIs)介导的,因此它们成为有吸引力的治疗靶点。在过去几年中,PPI接口被认为是不可挽回的,因为它们缺少口袋并且通常是扁平的,大的并且没有特征。然而,这种印象随着成功的PPI抑制剂而消失,并且药物基因组扩展了这些额外的靶标。 Benzodiazepines,ICG-001抑制b-连环蛋白和CBP之间相互作用,抑制剂IL2,MDM2,BCL-2 / BCL-XL,XIAP和VLA-4,)列为PPI靶向药物的实例。为了更好地解释阻断PPI的小分子的概念,图4显示了小分子对IL2和IL2R相互作用的阻碍,文献中还有其他已知PPI抑制剂的例子。可药用热点的主要特征是它们的位置偏好:通常在整个界面上没有观察到热点,而是聚集在一起,并且可药物热点倾向于位于互补口袋(蛋白质蛋白结合后消失的口袋)

图4.破坏IL2和IL2R之间PPI的小分子。 在右图中,IL2和IL2R(PDB ID:1Z92)之间的复合物,和小分子与IL2(PDB ID:1M49)之间的蛋白蛋白相互作用被叠加。 这种小分子阻断IL2与其受体之间的相互作用。 从左图中可以看出,小分子以红色显示的一些IL2-IL2R界面热点附近结合。 IL2-IL2R接口的热点是通过Hotpoint获得的。
变构药物(Allosteric drugs),即与PPI界面结合的药物,也能够改变PPI。 Ma等人建议设计针对变构热点的药物可能会以类似于可药用热点的方式扩大药物基因组; 并且开发识别变构热点的方法可以使变构药物设计更有效。 他们观察到Ras蛋白的变构区域中的热点,并声称Ras蛋白构象的变构调节可能是靶向Ras途径的有用方法。
七、热点和药物发现
小分子需要将其表面的一部分浸没到靶蛋白中以进行高亲和力结合。 通常,这通过与靶蛋白的腔(cavities)结合来完成(Wells和McClendon,2007)。 然而,蛋白质蛋白质相互作用(PPIs)是缺乏这样的空洞的治疗应用的有趣目标。 在PPIs上,结合能由热点控制,并且它们被其他残基埋在表面上(Moreira等,2007b)。 这一事实构成了一种有前途的蛋白质药物设计策略的基础,即热点移植(hot spot grafting)。
热点嫁接是从包含热点的文库中寻找合适的支架,并将这些热点浸没在表面上(Zhang和Lai,2011)。 基于图论的模式匹配方法或集合减少算法,可以用于定位适应期望的热点模式的支架(Liang等人,2000)。 Lai和他的同事开发了这样的算法,他们成功地设计了一种结合人EPO受体的蛋白质(Liu et al。,2007)。 Fleishman等人(Fleishman等,2011)提出了另一种设计具有预定热点模式的蛋白质药物的方法。 在他们的方法中,首先将支架蛋白停靠在靶蛋白上以找到具有形状互补的良好构象,然后使用热点图案来固定构型。 他们能够设计两种靶向流感血凝素保守stem区的蛋白质。
另一种基于热点引导结构的方法,HS-Pharm,由Barillari等人开发用于生物活性化合物设计。 HS-Pharm是一种基于知识的快速方法,通过训练具有已知配体结合口袋的机器学习算法,可以优先确定应该作为配体结合的空穴原子。 这里,热点用于聚焦基于结构的药效团模型。
设计针对热点的药物是打破PPI的良好策略,因为它们对稳定性有重大贡献。 此外,PPI的热点有助于检测配体高亲和力结合的区域。 PPI热点和配体结合热点之间的关系仍然受到质疑,但有一些研究指出它们之间的相关性。 受此事实的启发,Koes和Camacho(2012)提出了一种结构生物信息学方法,用于检测针对小分子设计的合理PPI的起点。 他们确定了界面残基簇,它们称为小分子抑制剂起始点(SMISP)。 这些区域比热点大,比整个界面小。
Seco等人(2009)合并了热点的识别和结合亲和力计算,以产生药物可靠性指数以发现可药用部位。他们的方法独立于表面描述符或训练集,它适用于PPI以及变构结合位点。他们在五种药理学靶点上测试了他们的方法,包括蛋白磷酸酶1B(PTP-1B),这是一个非常具有挑战性的目标。他们成功地鉴定了该蛋白质上的可药物部位,并发现最有效的可药物部位与PTP-1B和胰岛素受体酪氨酸激酶(IRK)界面重叠。李等人(2011)描述了使用多配体同时对接(multiple ligand simultaneous docking,MLSD)和热点的新型药物发现和药物重新定位方法。他们使用癌症靶标STAT3作为测试案例。首先,他们将多个药物支架对接到STAT3的热点并产生虚拟模板化合物。接下来,他们在药物数据库中搜索了类似的命中,并将塞来昔布(celecoxib)鉴定为STAT3的新型抑制剂。此外,他们根据一种先导模板和celecoxib模拟了两种新型先导抑制剂。
热点对于基于片段的药物发现也是至关重要的。众所周知,不同的探针仅聚集在热点周围(Hall等,2012)。 Hadjuk等在片段大小的化合物(Hajduk等,2005)和Liepinsh and Otting (1997)的实验中,通过使用核磁共振方法研究了水溶液中的有机溶剂。他们观察到两种分子都与热点结合,命中率为90%或大于90%。许多其他实验也得出了类似的结论,并证实热点能够结合一系列不同种类的小分子。 FTMap(Hall等,2012)发现了绑定热点,并为基于片段的药物发现提供了补充信息。 Kozakov等人提出了一种基于片段的方法,该方法即使在蛋白质处于其未结合状态时也起作用,以发现能够基于热点结合药物大小配体的蛋白质蛋白质界面上的区域。
蛋白质家族中的序列和结构相似性是药物发现中的最大挑战之一。 为了防止毒理学风险增加,应避免使用靶向保守的界面。
总之,热点区分PPI结合位点与蛋白质表面的其余部分,如前所述,它们构成了PPI结合能的大部分。 由于热点的调节成为阻断PPI的重要策略,这一发现对药物发现有很大影响。 它导致药物基因组的扩大和上面讨论的新药开发技术的出现。
八、热点和致病突变
在过去的十年中,由于1000国际基因组(Genomes Project et al。,2012),TCGA(癌症基因组图谱研究等,2013),HAPmap等大型项目的国际努力,已经产生了大量的序列数据。 同时,蛋白质数据库(Berman et al。,2007)中结构数据的积累继续增加。将蛋白质结构和遗传变异融合在一起,使研究人员能够更广泛地了解复杂的疾病机制。 2008年,出版了一套策划的PPI相关突变(Schuster-Bockler和Bateman,2008)。 之后,钟等人(2009)发表了一项关于不同类型疾病突变(节点清除和编辑扰动)的不同结果的开创性研究。随后,大卫等人(2012)声称致病突变更喜欢发生在给定蛋白质的界面区域,如果它们不在核心中。据了解,引起疾病的突变倾向于通过核心突变使其不稳定或通过界面突变改变其相互作用来改变靶蛋白的功能。同样,Wang等人(2012b)观察到框内突变富集在与相应疾病相关的蛋白质的相互作用界面区域中。
突变热点的不稳定效应,很可能发生在将引起疾病的突变与PPI联系起来的位置。尽管目前还没有针对这一主题的大规模研究,但一些研究人员发表了相关的案例研究。 Engin等(2013)模拟了ELANE和CSF3之间的相互作用。他们声称,101个残基的变体,即ELANE上的相互作用热点,可能正在改变CSF3-ELANE结合能的亲和力。此外,他们提到这些变异易于与乳腺癌患者的转移进展相关联。 Acuner Ozbabacan等(2014),研究了IL-1信号通路蛋白上的致癌突变和SNP的分布。他们能够将这些遗传变异中的一些映射到IL-1信号传导途径中相互作用的计算热点。后来,对IL-10与其他信号分子的结构相互作用进行了类似的研究,以了解致癌突变对导致癌症和炎症的机制的影响(Acuner-Ozbabacan等)。
九、热点和翻译后修改
翻译后修饰(Post-translational modifications,PTM)辅助PPI,同时细胞响应各种动态信号。在功能性位点发生的PTM事件能够创建新的结合位点(Schaller和Parsons,1995),增加蛋白质的相互作用伙伴数量,从而使功能多样化。同时,PTM也可能导致结合伴侣的解离(Nussinov等,2012)。已经表明磷酸化位点更可能发生在异源寡聚和弱瞬时同源寡聚体复合物中的结合界面上,而结合热点倾向于在异源寡聚体中磷酸化。此外,据称磷酸化能够引起一些复合物的结合能的显着变化(Nishi等,2011)。同一研究的另一个结论是,与异构寡聚体界面中的其余残基相比,界面磷酸盐结合热点的可能性是其两倍。
十、结论
- 蛋白质蛋白质相互作用的结合亲和力和特异性由能量上重要的残基提供。
- 这些残基的突变导致蛋白质的解离或迫使它们改变它们的结合模式。
- 这些能量上有利的残基(热点)也是蛋白质蛋白质上的药物样分子界面的残基。 对于药物开发研究,指定热点残基作为目标位点将指导设计这些特定残基的小分子。 该策略可以防止药物可能的副作用。
- 通过实验找到这些热点残基是一个昂贵的过程,这使得研究人员开发出有效预测热点的计算算法。 过去十年中热点预测的改进是有希望的,并且将这些研究与药物发现相关联对未来是有希望的。
参考资料
- Progress in Biophysics and Molecular Biology (2014) 《Hot spots in proteineprotein interfaces: Towards drug discovery》 http://home.ku.edu.tr/~okeskin/Engin_Billur_HotspotsReview.pdf
