【10.1】单氨基酸于疾病关联预测-SAPRED
Classifier-based methods:SAPRED
Single Amion acid Polymorphisms disease association Predictor,把问题转化成统计上的supervise的classification这样的一个问题.classification就是根据你有的一些知识,通过已知的数据,新拿到一个数据。基于已知的一些遗传变异会导致疾病,还有一些它不会导致疾病,然后我们来预测一些新的遗传变异是否会导致疾病,这就是一个supervised classification的问题。这个统计上有很多非常重要的方法你可以用来使用,包括比较简单的比如线性回归,logistic regression,naive bayes。而该算法采用的是一个支持向量机,也是一个很强大的统计方法。
在做之前,需要定义你针对每一个遗传变异,它是什么样的一个特征,那么我们就定义序列和结构上的特征。跟polypen一样,我们要看这个蛋白有没有已知的三维结构,已知的三维结构主要存储在PDB这样的一个protein data bank这个数据集里。如果你感兴趣的蛋白没有已知的结构就可以试试看能不能同源建模,同源建模就是要找到和你蛋白类似的,结构已知的蛋白,通过你已有的序列比对,已知蛋白有这样的已知结构,那这个蛋白和它序列上做比对的话,那么能比对上的位点就可以借鉴已知蛋白的这些三维上的这些坐标,然后再用能力方程把比对不上的这些位点和改变了的那些氨基酸通过一个能量方程,把它位置给定下来。我们用这个方法先拿到一个可用的结构。
我们定义特征就和别人的是一样的,包括这个conservation(评估的这个位点整体上来看是很保守的还是不保守的,我不管具体它是从那个氨基酸到哪个氨基酸),residue frequencies(评估的是每一种氨基酸在一个位点在演化上出现的频率),这是有关联性的两个特征,但侧重点不太一样。
在结果上,该方法沿用了别人用的,到底在蛋白内部还是表面,在三维结果什么位置上,在二级结构的什么位置上。
- 定义了一些新的特征,其中一个呢,再后来分析中被证明还是很有效的一个特征,叫做structrual neighbor profile,有一些结构上的位点,其实并不是说这个氨基酸本身有什么最重要,可能就是这样的位点它周围的一个微环境,它周围存在的这些氨基酸,可能就是导致它存在的这些位点是不能随便改变的,只要这个位点改变,它整个影响结构的概率是比较高,在你比较感兴趣的这样一个为带你,在周围画它一个球体,看中间这20种氨基酸每一样都有多少个。这个球画多大呢,不同半径和它预测能力是不一样的,最终显示13个Angstroms是预测能力最强的一个半径
- 第二个特征,在之前方法你会看就是你发现的这个位点它会不会是刚好在一个功能区间上,但其实你想像一下,真正能落在功能区间的active site上的位点其实还是很少的这样的一个蛋白,但是以我们的知识来看,它在这个绝对的active site附件的很多氨基酸其实的改变也会影响这个功能,也会影响它和其他分子的结合,所以我们就定义了一个所谓的nearby functional sites这样的一个特征,它不光是考虑是否刚好落在active site,同时也看变异发生离它最近的一个active site的距离是多少,而这个距离可以从序列和结构两个层次来定义的。
- 第三个特征,PDB里的蛋白有一些结构的一些位置是没有三维坐标的,其中很多就是因为它是一个无序的结构,它本来在自然情况下是摆来摆去的,就是没有一个三维的坐标位置。那么这些disorder的region额,这些位置可能它如果有一个变异无所谓,本来这些结构就不固定,但是,当集训集里有这样的122个氨基酸的改变处在disorder region,但是其中144个,也就是93%都是会导致疾病的,所以这些disorder的无序区间反而可能会对维持它的wild type的氨基酸反而很重要,反而更加不能随便的改变。
- 第四个特征, 计算了氢键的改变,因为你替换了别的氨基酸,它和周围的氨基酸能形成氢键的数目就会有改变,这个就是比较直观的,就是你这个氢键的改变越多,致病的概率就会越大
- 第五个特征, 当时数据集里有52个跨膜区的变异,其中94%跨膜区的变异都会导致疾病的;其中194个会影响蛋白聚集变异(87%是致病的),就是说如果一个突变它是影响了蛋白聚集的能力,那么它最后对表型的影响是比较大的,还有一个就是反的方向,如果它放生在HLA这个家族里,那么435个里有434个是不会影响最终表型的
把所有的信息做一个统计(feature selection),选出来的其中和你要预测的事情它关联最大的那一组,然后用一个支持向量机的方法,那怎样评估它的准确度呢,比较常用一个方法就是叫做five-fold cross-validation,把你的这个随机分成5份,然后选出其中的一份,用另外四份做一个预测模型,然后再取出来的一份来看,你预测的准确度有多少,然后拿出来另外一份,看看你的这个结果怎么样。
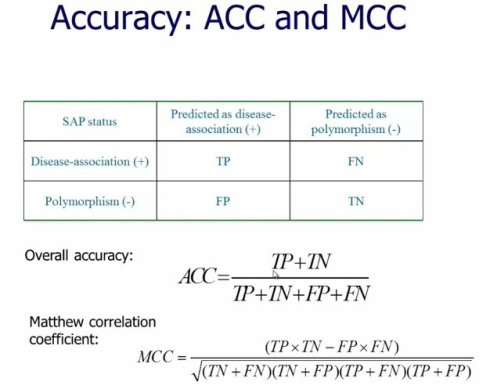
如果评估一个准确度,那么就是TP+TN除以所有的数量 评估的结果显示residue frequencies,从序列上,演化上这个位置看到那些氨基酸仍然是预测能力最强的特征
参考资料
北大魏丽萍老师的课件
