【6.7.3】Digenome-seq
(体外的方法,通过测序评估脱靶情况)
摘要
尽管现在通过CRISPR-Cas9系统进行RNA指导的基因组编辑已广泛用于生物医学研究,但Cas9核酸酶的全基因组靶标特异性仍存在争议。在这里,我们介绍Digenome-seq,体外Cas9消化的全基因组测序,以分析人细胞中全基因组的Cas9脱靶效应。该体外消化产生可在计算上鉴定的切割位点具有相同5’端的序列读数。我们验证了脱靶位点,在该位点处插入或缺失的频率低于0.1%,接近目标深度测序的检测极限。我们还表明,Cas9核酸酶可以是高度特异性的,仅在整个基因组中的几个(而不是数千)位点诱导脱靶突变,并且可以通过和修饰的sgRNA替换“混杂promiscuous”单向导RNA(sgRNAs)来避免Cas9脱靶效。 Digenome-seq是一种功能强大,灵敏,无偏且具有成本效益的方法,可用于分析包括Cas9在内的可编程核酸酶的全基因组脱靶效应。
一、主体
可编程核酸酶,包括锌指核酸酶,转录激活子样效应核酸酶和RNA引导的工程核酸酶(RGENs, RNA-guided engineered nucleases)。RGENs源自II型簇状,规则间隔,短回文重复序列(CRISPR)-CRISPR相关(Cas)原核适应性免疫系统,现已广泛用于培养细胞和整个生物体的基因组编辑。不幸的是,这些核酸酶可以诱导脱靶突变。例如,RGEN-由化脓性链球菌和sgRNA衍生的Cas9蛋白组成,可识别由20 bp序列组成的22 bp靶DNA序列,该序列与sgRNA和5'-NGG-3’原型间隔子杂交, Cas9识别的邻近基序(PAM)序列可以耐受多达几个核苷酸位置的错配。这表明在人类基因组中,给定核酸酶的脱靶位点多达数千个。脱靶DNA切割可导致意外的基因组位点发生突变,并导致染色体重排,例如易位,缺失和倒位;这种影响引起人们对该技术在研究和医学中的应用的关注。
据报道,有多种策略可降低RGEN的脱靶效应,包括使用在5’端带有两个额外鸟嘌呤核苷酸的sgRNA,截短的sgRNAs,成对的Cas9切口酶和催化死亡的Cas9(dCas9)-FokI融合蛋白,以及纯化的Cas9蛋白的递送。尽管已显示出这些不同的方法可以在多个位点将脱靶突变的频率降低至少一个数量级,但仍不清楚这些RGEN变体在整个基因组中是否没有脱靶效应。为了解决这个关键问题,研究人员必须开发出以无偏见的方式在基因组规模上询问RGENs特异性的方法。
在这项研究中,我们使用全基因组测序(WGS)来分析使用RGEN创建的克隆基因敲除(KO)细胞中的全基因组RGEN脱靶突变。 此外,我们通过对核酸酶消化的基因组DNA进行测序,在大量细胞中鉴定了RGEN脱靶位点。 我们的结果表明,RGENs具有高度的特异性,仅在整个基因组中的少数(而非数千)位点诱导脱靶突变,并且通过使用修饰的sgRNA可以避免这些脱靶效应。
三、结果
3.1 人单倍体细胞的全基因组测序
我们选择了人类单倍体细胞系HAP1(参考资料33)来分离携带RGEN诱导的突变的基因KO细胞的克隆种群。在单倍体细胞中,相同数目的读数可转化为两倍于二倍体细胞的测序深度,并且过滤器可以去除杂合变体。我们分离了五个不同的KO HAP1细胞系,每个细胞系在激酶基因(ABL1,EPHB2,ERBB3,FGFR2和FGFR4;补充图1)中都有一个单一的破坏。从这些细胞和野生型细胞中分离出的基因组DNA均经过WGS处理。为了测试WGS的可重复性,我们对野生型细胞和ABL1-细胞一式两份进行了测序。
我们使用了变体调用程序Isaac来识别相对于hg19参考基因组(图1)的小插入或缺失(indels)而不是点突变(图1),因为可编程核酸酶很少产生取代。应用生物信息学过滤器后,我们获得了每个基因组中唯一的2,026–3,250个插入缺失(补充表1和补充说明1)。然后,我们将RGEN目标位点与这些插入/缺失位置进行了比较。只有9–84个插入缺失位点包含一个PAM序列,并且与各自的靶序列至少有10个核苷酸匹配(补充表1)。这些插入/缺失中只有一个通过Sanger测序验证(补充图2a),并且是由自发突变引起的(补充图2b)。我们用Isaac或Integrative Genomics Viewer(IGV)35(补充图2c,d)确认了所有五个靶向突变。
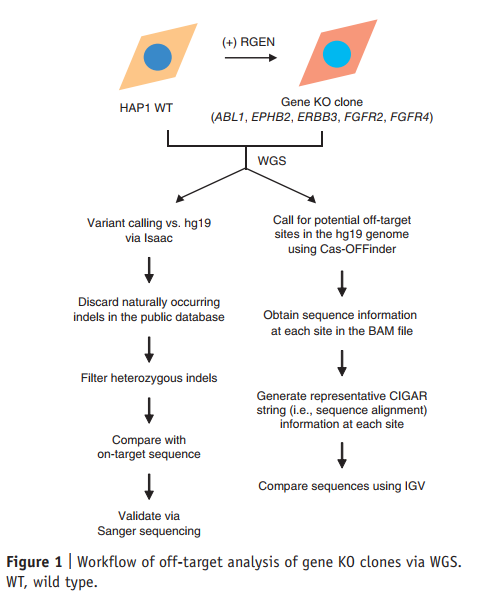
3.2 检查潜在的脱靶站点
接下来,我们使用Cas-OFFinder36( http://www.rgenome.net )及其升级版本列出了100,000个以上的同源位点,这些位点与目标位点的差异最大为8 nt,或者与DNA或DNA的差异最大为2 nt。 RNA凸起(补充图3a),我们检查了每个基因组序列中这些潜在的脱靶位点是否存在核酸酶诱导的插入缺失。我们开发了一种计算机程序,以将在> 100,000个同源位点附近对齐的序列读数与参考序列进行比较(补充图3b)。值得注意的是,该程序成功地鉴定了五个KO克隆中的所有五个靶向突变(补充图3c)。仍然没有发现脱靶插入缺失。
这些结果表明,RGEN具有高度的特异性,在单细胞来源的克隆中不存在脱靶突变。但是,无法通过分析几个克隆来检测大量细胞中频率低于10%的脱靶突变,这可能在基因和细胞治疗应用中引起问题。此外,RGENs是否可以在成百上千个脱靶位点诱导低频插入缺失仍是未知的。
3.3 Digenome-seq:直线与交错排列 straight versus staggered alignment
我们认为,我们可以通过对体外核酸酶消化的基因组(二糖体)进行测序来鉴定大量细胞中RGEN诱导的脱靶突变。 这些消化物应产生许多具有相同5’端的DNA片段,从而产生在切割位点垂直排列的序列读数。 相反,所有其他序列读取将以交错方式进行比对。
我们选择了HBB基因特异的RGEN,该基因已被证明在高度同源位点(称为OT1位点)上引起脱靶突变。 我们还分析了三个其他同源位点(OT3,OT7和OT12),它们与目标位点相距3 nt。 该RGEN在基因组背景下在体内和体外有效地切割了靶标,OT1和OT3位点(图2和补充说明2)。
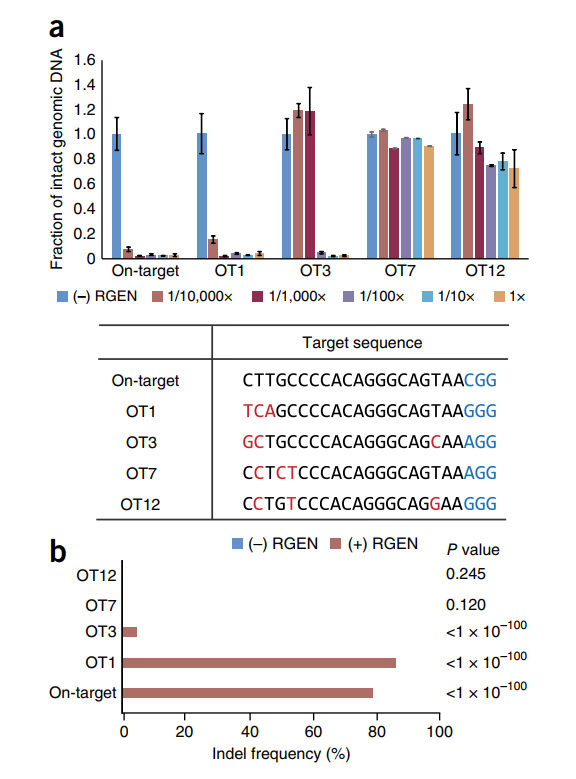

使用WGS对四组不同的基因组DNA进行了研究,以研究使用RGEN进行的基因组DNA体外消化是否可以产生在切割位点处具有直接比对的序列读数。 分离自RGEN和模拟转染的HAP1细胞的基因组DNA在体外用300 nM的HBB RGEN消化,以确保完全裂解。 平行地,将从转染和模拟转染的细胞中分离的完整基因组DNA进行WGS,无需体外RGEN消化(图3a)。 在将序列读图映射到参考基因组后,我们使用IGV观察了靶位和四个同源位点的序列比对模式。
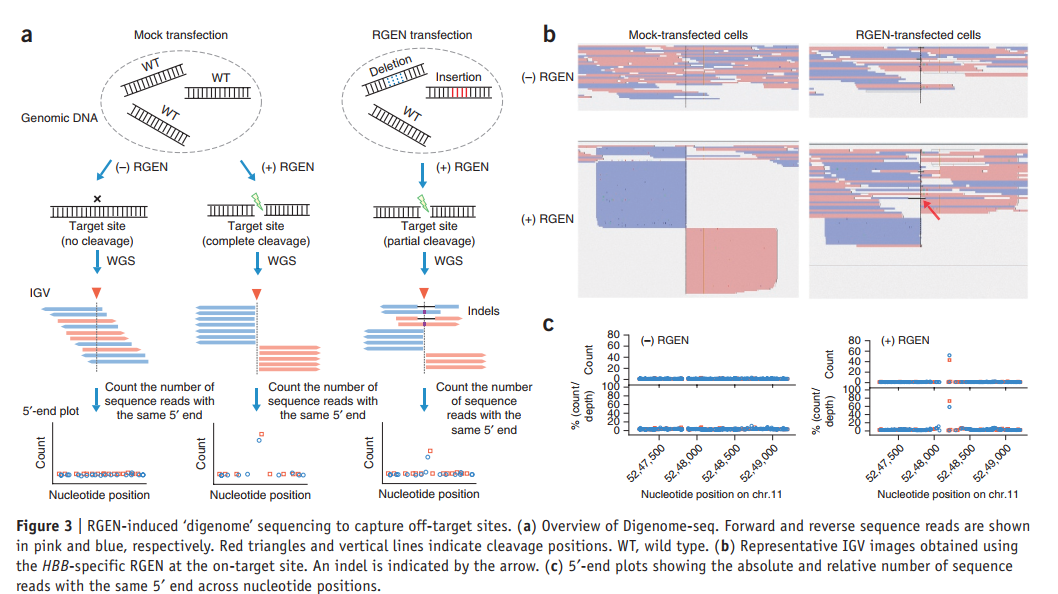
首先,我们检查了从模拟转染细胞中分离的双基因组。在目标上OT1和OT3的位置,观察到不同寻常的直线排列模式(图3b和补充图4a,b)。跨越切割位点的序列读数非常罕见。在没有RGEN处理的情况下,没有观察到这样的直线比对。在OT7和OT12位点,大多数序列读数跨越了潜在的切割位点(PAM上游3 bp),导致错开比对(补充图4c,d)。
其次,我们比较了从RGEN转染的细胞中分离的双基因组与各自完整的基因组。在所有五个位点,完整的基因组产生了交错排列的典型模式(图3b和补充图4)。相反,双基因组在靶上和OT1位点均显示出直线和交错排列。在这两个位点,几乎所有重叠的序列读数都导致包含插入缺失的错位比对。显然,这些插入/缺失序列在体外没有被RGEN切割。未发现跨越OT7和OT12位点的重叠序列读取的插入缺失。 OT3位点是一个有趣的案例:双基因组显示了一个带有几个重叠序列读数的笔直模式。值得注意的是,一个重叠序列包含一个插入缺失,最可能是由RGEN诱导的(补充图4b)。这些结果表明,Digenome-seq足够灵敏,可以鉴定罕见的脱靶突变,并且序列读数的垂直或垂直比对是RGEN体外裂解的唯一特征,尽管并非所有具有直接比对的位点都是真正的偏离-目标sites。
3.3 基因组规模的脱靶位点
我们开发了一种计算机程序,用于搜索整个基因组中序列读数的直接比对。首先,我们绘制了其5’末端开始于HBB靶上核苷酸附近的核苷酸位置和两个经过验证的靶外位点(单核苷酸分辨率)的序列读数的计数(补充图5a)。我们假设应该在切割位点彼此相邻观察到几乎相等数量的序列读数,分别对应于Watson或Crick链,产生双峰。正如预期的那样,双基因组在三个切割位点产生了双峰(图3c和补充图5b,c)。未在体外进行RGEN处理的完整基因组在这些位点未产生此类双峰模式。
接下来,我们将这种方法应用于整个RGEN转染的二基因组,模拟转染的二基因组,完整的RGEN转染的基因组和完整的模拟转染的基因组。此外,模拟转染的基因组DNA在不存在sgRNA的情况下在体外用Cas9蛋白处理,或者在RGEN(3 nM Cas9)浓度低100倍的条件下进行了处理,然后进行了WGS和二基因组分析。我们以计算方式搜索了两条链中具有相同5’末端的序列读数计数均大于10的位点,并且其中至少20%的序列读数垂直对齐。在分别用3 nM和300 nM RGEN处理的模拟转染的基因组中,总共鉴定出17个和78个位点,包括靶上和两个经过验证的靶外位点(图4a),显示双峰5’端图中的图形和IGV图像中的直线对齐。在RGEN转染的基因组中,我们在125个位点观察到了这种模式,包括三个经过验证的靶位和离靶位点。 OT7和OT12位点在这三个基因组中未显示双峰模式。通常在这三个双基因组中鉴定出大多数位点,证明了双基因组-seq的高再现性。因此,在模拟转染的二基因组(3 nM RGEN)中发现的16个候选位点(不包括一个假阳性位点)中的15个(94%)也在其他两个独立的二基因组中被鉴定出来(图4a)。但是在122个位点(不包括3个经过验证的位点)中,没有一个在RGEN转染的二基因组中伴随插入缺失,这表明这些候选位点的突变很少发生,甚至根本没有发生。在三个完整基因组中只有几个位置观察到了这种双峰模式(补充表2)。在完整基因组中鉴定出的所有这些位置均为假阳性,这是由于相对于参考基因组,HAP1基因组中天然存在的插入缺失所致(补充图6)。因此,双峰模式或序列读数的直接比对是在三个二基因组中发现的独特特征。


我们比较了在RGEN转染和模拟转染的二基因组中鉴定的74个常见位点的DNA序列与20 bp的靶向位点,发现在20 nt的核苷酸中,除5’末端的一个均保守(图4b)。有趣的是,sgRNA中的5’碱基是错配的鸟嘌呤,在U6启动子的控制下转录是必需的。靶序列中的5’碱基是胞嘧啶。此外,通过将74个位点的DNA序列相互比较而不是与靶上序列进行比较而从计算上获得的从头基序清楚地显示了除前2 nt以外的所有位置与靶上序列的匹配(图4c)。值得注意的是,在这些双峰位置中有70个(95%)伴有5'-NGG-3’PAM,恰好位于预期裂解位点下游3 nt。通过允许DNA / RNA凸起或将5'-NGA-3’或5'-NAG-3’假定为非规范PAM,将某些位点与靶位点相匹配。其他位点与目标序列没有明显的序列同源性,表明它们是假阳性。
我们还发现,同源位点的错配越少,被Digenome-seq捕获的可能性就越大。因此,在157个同源位点中,有21个(13%)与目标位点相差3或4 nt,但在1,191个位点中只有15个(1.2%),在7,896个位点中只有1个(0.013%)分别捕获了5 nt和6 nt的差异(图4d)。综上所述,这些结果表明,大多数双峰模式是由体外RGEN消化引起的,并且Digenome-seq可以在基因组背景下捕获核酸酶切割位点。
3.4 验证候选site上的脱靶效应
我们进行了靶向深度测序,以验证或使两个独立的双基因组中鉴定的74个常见位点的脱靶效应无效(图4e和补充表3)。另外,我们测试了与目标位点相差3 nt但未被Digenome-seq捕获的其他八个位点。在这八个位点均未检测到偏离目标的插入缺失,其频率至少为0.1%,并且大于阴性对照的频率(Fisher精确检验,P <0.01)(图4d和补充表3)。在这74个位点中的五个位点(包括已验证的靶点,OT1和OT3位点)观察到插入缺失,频率介于0.11%至87%之间(图4e和补充图7)。在另两个新验证的脱靶位点(称为HBB_48和HBB_75)上,检测到插入缺失的频率分别为0.11%和2.2%。这两个站点与目标站点的差异为3 nt。相对于20-nt sgRNA序列,在HBB_48位点有3个核苷酸错配,在HBB_75位点有2个错配,在5’末端与靶点位点相差1 nt。与20-nt sgRNA序列相比,这些经过验证的脱靶位点都没有DNA / RNA凸起,也没有非典型PAM(5'-NGA-3’或5'-NAG-3')。请注意,这两个新的脱靶位点和其他三个位点分别在三个二基因组中的每一个中捕获,说明了Digenome-seq组序列的高灵敏度和可重复性。
3.5 Digenome-seq与另一个“混杂”(promiscuous) RGEN
我们用另一种RGEN进行了Digenome-seq测序,已证明其可诱导VEGFA基因座上的靶上突变和四个同源位点上的脱靶突变。总共捕获了81个站点,包括目标上的站点和四个经过验证的目标外站点,这些站点显示出双峰模式(补充图8和9)。在这81个位点的所有DNA序列都包含规范的5'-NGG-3’PAM。这些序列与靶上序列的比较表明在每个核苷酸位置上都匹配。我们还对这些序列进行了比较,从而获得了从头序列:所得到的序列徽标还显示出几乎在每个核苷酸位置上都与靶序列相匹配,这表明20-nt sgRNA序列中的每个核苷酸都有助于特异性(补充图8b,c)。
然后,我们使用靶向深度测序来确认在通过二基因组分析捕获的81个位点和与目标上的位点相差3个或更少核苷酸但未被捕获的28个位点上的靶上和脱靶效果。该RGEN在目标位点和先前验证的四个脱靶位点产生插入缺失,频率范围为0.32%至87%。此外,还验证了通过Digenome-seq捕获的四个新的脱靶位点,在该位点以0.065%±0.021%到6.4%±1.2%(±sem)的频率诱导插入缺失(补充图8e和图8e。 10)。这些脱靶位点与20-nt靶序列含有1-6个核苷酸错配,并且在PAM近端种子区域中至少有一个错配。人类基因组中有13 892个6 nt错配的位点,但Digenome-seq仅捕获了6个位点(0.043%)。其中,只有一个位点通过深度测序得到了验证(补充图8d,e)。尽管通过Digenome-seq捕获的81个位点中有40个位点与20-nt靶标序列相比缺失或多余,但这些经过验证的脱靶位点均未包含DNA / RNA凸起。在所有其他位点,包括未被Digenome-seq捕获的位点,插入缺失频率均低于0.05%,或小于或没有统计学上与使用空载体对照获得的频率不同。
3.6 通过修饰的sgRNA避免RGEN脱靶效应
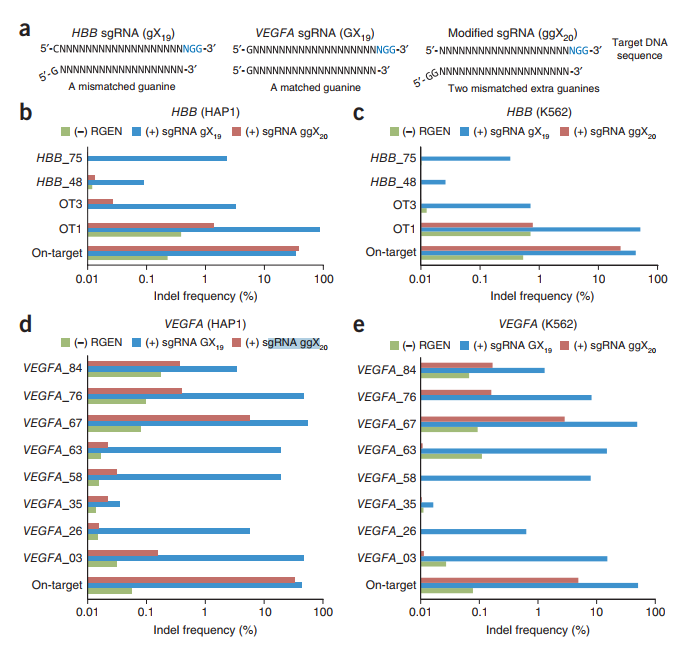
在5’末端带有两个额外鸟嘌呤的sgRNA(称为ggX20 sgRNA)可以有效地区分靶位与同源位点相差≥2 nt的位点,从而在不牺牲靶位效应的情况下将脱靶效应降低了几个数量级。我们用各自的ggX20 sgRNA(“ g”和“ G”分别代表不匹配的鸟嘌呤和匹配的鸟嘌呤)替换了两个混杂的gX19(HBB)和GX19(VEGFA)sgRNA(图5a),并进行了靶上和脱靶测量HAP1和K562(人类白血病)细胞中的目标突变频率已验证位点。令人惊讶的是,当我们使用ggX20 sgRNA时,在两个基因中几个脱靶位点的深度测序错误率以上都几乎检测不到插入缺失(图5b-e和补充表4)。与gX19和GX19 sgRNA相比,这些sgRNA在HAP1细胞的各个靶位上的活性几乎相同,尽管它们在K562细胞的靶位上的活性不如两个常规sgRNA。根据靶上与靶外插入缺失频率的特异性比,两种修饰的sgRNA的特异性比常规sgRNA高660倍(补充表5)。

四、讨论区
CRISPR-Cas系统的全基因组特异性在RNA引导的基因组编辑领域受到广泛关注。但是,关于RGEN特异性的报道看似矛盾的结果。我们首先使用了T7E1分析,然后使用了深度测序来显示,即使在人类细胞中与目标位点相距2 nt的位点,RGEN都不会留下偏离目标的足迹。与这些结果一致,全外显子组测序和WGS3均表明,在克隆的细胞群体中,RGEN在整个人类外显子组和基因组中分别没有诱导脱靶插入缺失。与之形成鲜明对比的是,其他几个研究小组报告说,RGENs可以在相距最多5 nt的位点诱导脱靶插入缺失(参考文献17,18,19,20,21),或者与靶标相比缺少或包含额外的核苷酸序列。这些结果表明,给定的核酸酶可能识别并切割基因组中多达数千个脱靶位点。一个接一个地测量所有这些潜在站点的脱靶效应几乎是不可能的任务。 RGENs可能在成百上千个脱靶位点诱导插入缺失,其频率分别低于1%或0.1%,而仅对少数几个克隆进行测序就无法检测到。 sgRNA也可能具有广泛不同的特异性:某些可能具有高度特异性,而另一些则混杂。为了解决这些问题,我们选择了两种混杂的sgRNA,其他人已经证明它们可以诱导人细胞中的高频脱靶诱变,并通过Digenome-seq检查了它们在全基因组范围内的脱靶效应。
为了以无偏倚的方式分析工程化核酸酶的全基因组脱靶效应,已使用SELEX(通过指数富集的配体系统进化)和染色质免疫沉淀测序[41,42,43]。这些方法依赖于DNA结合而不是DNA裂解。不幸的是,这些事件通常是不相关的:大多数由dCas9识别的脱靶DNA结合位点根本不会被细胞中的Cas9裂解。整合酶缺陷型慢病毒载体的捕获和体外选择是检测核酸酶切割位点而不是结合位点的两种不同方法。这两种方法是互补的,但都不是全面的。因为在给定位点捕获IDLV的效率远低于该位点的突变频率,所以该方法无法捕获许多真正的脱靶位点。体外选择使用RGEN裂解包含10^12个以上变异体的偏向DNA底物文库,并通过深度测序确定裂解的文库成员。不幸的是,大多数切割的序列在基因组中不存在。此外,这种方法无法检查可以与DNA / RNA凸起杂交的序列的脱靶切割。
在这里,我们证明了Digenome-seq是一种可再现且灵敏的方法,可以无偏倚地分析核酸酶脱靶效应。
- 首先,Digenome-seq依赖于DNA切割而不是结合。
- 其次,与体外选择方法不同,Digenome-seq在基因组环境中进行。值得注意的是,Digenome-seq捕获了带有DNA / RNA凸起的潜在脱靶位点。
- 第三,Digenome-seq足够灵敏,可以检测到以0.1%或更低的频率诱导插入缺失的脱靶位点,接近高通量测序平台的检测极限。
- 第四,Digenome-seq具有成本效益:借助Illumina HiSeq X 10,WGS现在的价格为1,000美元。
- 最后,该方法是可重现的,如三个独立的二基因组分析所示。
最近,有两个小组报道了称为HTGTS和GUIDE-seq的方法,用于捕获Cas9在细胞中诱导的双链断裂(DSB)。我们注意到在这些研究中也分析了VEGFA sgRNA。有趣的是,HTGTS和GUIDE-seq也捕获了由Digenome-seq识别的9个经过验证的靶点和脱靶位点中的8个,表明这三种方法产生了可比的结果。与HTGTS和GUIDE-seq不同,Digenome-seq不受染色质可及性的限制。此外,HTGTS和GUIDE-seq要求
- 滤除与目标位点同源性差的捕获位点,以丢弃由于细胞中随机发生的DSB或PCR伪像而产生的许多假阳性位点,
- 搜索同源位点由于细胞中DSB修复伴随着可变长度的插入/缺失,因此在捕获位点周围
这两个因素都会带来偏差。
在这项研究中,我们将Digenome-seq应用于常规的Cas9,但该方法还应该分析由dCas9-FokI,成对的Cas9切口酶或其他可编程核酸酶引起的全基因组脱靶效应。在将此类核酸酶用于基因或细胞治疗应用之前,可以通过Digenome-seq仔细监测其脱靶效应,以避免不必要的突变。该方法还将在下一代基因组编辑工具的开发中提供试金石。
五、方法
试验略。。
用于测序数据分析的流程:
https://github.com/chizksh/digenome-toolkit2
分析同源位点的脱靶效应。
我们使用Cas-OFFinder( http://www.rgenome.net )查找潜在的脱靶位点,这些位点与脱靶序列相差最多8 nt,而从1到5则相差最多2 nt。 nt DNA或RNA凸起。 接下来,我们从BAM文件中潜在的切割位点获得了±10 bp左右的CIGAR字符串信息,并得出了最常见的CIGAR字符串。 接下来,我们将最常见的CIGAR字符串与野生型序列进行比较,以鉴定具有插入缺失的候选位点。 可根据要求提供本研究中使用的计算机程序。 我们使用IGV逐一验证或验证这些候选位点的插入缺失。
参考资料
- Kim, D., Bae, S., Park, J. et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods 12, 237–243 (2015). https://doi.org/10.1038/nmeth.3284
