【1.1】基因组注释
基因组注释主要包括四个研究方向:
- 重复序列的识别
- 非编码RNA的预测
- 基因结构预测
- 基因功能注释
一、基本概念
1.重复序列的识别
1.1.1 重复序列的研究背景和意义
重复序列可分为:
- 串联重复序列(Tendam repeat),包括有微卫星序列,小卫星序列等等;
- 散在重复序列(Interpersed repeat),散在重复序列又称转座子元件,包括以DNA-DNA方式转座的DNA转座子和反转录转座子(retrotransposon)。常见的反转录转座子类别有LTR,LINE和SINE等。
1.1.2 重复序列识别的发展现状
目前,识别重复序列和转座子的方法为
- 序列比对
- 从头预测两类。
序列比对方法一般采用Repeatmasker软件,识别与已知重复序列相似的序列,并对其进行分类。常用Repbase重复序列数据库。
从头预测方法则是利用重复序列或转座子自身的序列或结构特征构建从头预测算法或软件对序列进行识别。从头预测方法的优点在于能够根据转座子元件自身的结构特征进行预测,不依赖于已有的转座子数据库,能够发现未知的转座子元件。常见的从头预测方法有Recon,Piler,Repeatscout,LTR-finder,ReAS等等。
1.1.3 重复序列识别的研究内容
获得组装好的基因组序列后,我们首先预测基因组中的重复序列和转座子元件。
- 一方面,我们采用RepeatScout、LTR-finder、Tendem Repeat Finder、Repeatmoderler、Piler等从头预测软件预测重复序列。为了获得从头预测方法得到的重复序列的类别信息,我们把这些序列与Repbase数据库比对,将能够归类的重复序列进行分类。
- 另一方面,我们利用Repeatmasker识别与已知重复序列相似的重复序列或蛋白质序列。通过构建Repbase数据库在DNA水平和蛋白质水平的重复序列,Repeatmasker能够分别识别在DNA水平和蛋白质水平重复的序列,提高了识别率。
1.1.4 重复序列识别的关键技术难点
-
第二代测序技术测基因组,有成本低、速度快等优点。但是由于目前产生的读长(reads)较短。由于基因组序列采用kmer算法进行组装,高度相似的重复序列可能会被压缩到一起,影响对后续的重复序列识别。
-
某些高度重复的序列用现有的组装方法难以组装出来,成为未组装reads(unassembled reads)。有必要同时分析未组装reads以得到更为完整的重复序列分布图。之前,华大已开发了ReAS软件,专门用于识别未组装reads中的重复序列。但该软件目前只能处理传统测序技术(如sanger测序)生成的较长片段的reads,需要进一步改进方可用于分析第二代测序技术得到的reads。同时,未组装的短片段reads重复度更高,识别其重复区域具有较大难度。
1.1.5 重复序列识别的研究方向
- 整合现有的重复序列预测方法,对组装好的基因组序列进行分析。
- 综合考虑并结合短序列组装策略,校正重复序列识别的结果。
- 开发识别未组装reads重复序列的算法和流程并构建一致性序列。
1.2.非编码RNA序列的预测
1.2.1 非编码RNA预测的研究背景和意义
非编码RNA,指的是不被翻译成蛋白质的RNA,如tRNA, rRNA等,这些RNA不被翻译成蛋白质,但是具有重要的生物学功能。
- miRNA结合其靶向基因的mRNA序列结合,将mRNA降解或抑制其翻译成蛋白质,具有沉默基因的功能。
- tRNA (转运RNA)携带氨基酸进入核糖体,使之在mRNA指导下合成蛋白质。
- rRNA(核糖体RNA)与蛋白质结合形成核糖体,其功能是作为mRNA的支架,提供mRNA翻译成蛋白质的场所。
- snRNA(小核RNA)主要参与RNA前体的加工过程,是RNA剪切体的主要成分。
1.2.2 非编码RNA预测的发展现状
由于ncRNA种类繁多,特征各异,缺少编码蛋白质的基因所具有的典型特征,现有的ncRNA预测软件一般专注于搜索单一种类的ncRNA,如tRNAScan-SE 搜索tRNA、snoScan 搜索带C/D盒的snoRNAs、SnoGps 搜索带H/ACA 盒的snoRNAs、mirScan 搜索microRNA等等。Sanger实验室开发了Infernal软件,建立了1600多个RNA家族,并对每个家族建立了一致性二级结构和协方差模型,形成了Rfam数据库。采用Rfam数据库中的每个RNA的协方差模型,结合Infernal软件可以预测出已有RNA家族的新成员。Rfam/Infernal方法应用广泛,可以预测各种RNA家族成员,但是特异性较差。
我们建议:如果有更好的专门预测某一类非编码RNA的软件,那么采用该软件进行预测;否则,使用Rfam/Infernal流程。
1.2.3 非编码RNA预测的研究内容
利用Rfam家族的协方差模型,我们采用Rfam自带的Infernal软件预测miRNA和snRNA序列。由于rRNA的保守性很强,为此我们用序列比对已知的rRNA序列,识别基因组中的rRNA序列。tRNAscan-SE工具中综合了多个识别和分析程序,通过分析启动子元件的保守序列模式、tRNA二级结构的分析、转录控制元件分析和除去绝大多数假阳性的筛选过程,据称能识别99%的真tRNA基因。
1.2.4 非编码RNA预测中拟解决的关键技术难点
识别非编码RNA的假基因:基因组中很多序列由非编码RNA基因复制而来,与非编码RNA基因序列相似,但不具有非编码RNA的功能。目前我们采用的非编码RNA序列的预测方法都是基于序列比对和结构预测,不能够很好的去除这类非编码RNA的假基因。针对这个问题,我们考虑结合RNA表达信息如RNA-seq数据进行筛选。
1.2.5 非编码RNA预测的研究方向
- 专门检测小片段RNA序列的方法现在已经得到广泛应用,利用小片段RNA序列数据进行非编码RNA的预测是我们的重要研究方向。
- 开发miRNA靶向基因预测流程:miRNA通过调控其靶向基因的mRNA稳定性或翻译来控制生命活动的进程。预测miRNA靶向基因能够给我们研究miRNA功能带来提示。由于miRNA在动物和植物中对靶向基因的调控机制差别较大,我们建议对动物和植物分别建立靶向基因预测流程,提高预测准确度。
1.3 基因结构预测
1.3.1 基因结构预测的研究背景和意义
通过基因结构预测,我们能够获得基因组详细的基因分布和结构信息,也将为功能注释和进化分析工作提供重要的原料。基因结构预测包括预测基因组中的基因位点、开放性阅读框架(ORF)、翻译起始位点和终止位点、内含子和外显子区域、启动子、可变剪切位点以及蛋白质编码序列等等。
1.3.2 基因结构预测的发展现状
原核生物基因的各种信号位点(如启动子和终止子信号位点)特异性较强且容易识别,因此相应的基因预测方法已经基本成熟。Glimmer是应用最为广泛的原核生物基因结构预测软件,准确度高。而真核生物的基因预测工作的难度则大为增加。
- 首先,真核生物中的启动子和终止子等信号位点更为复杂,难以识别。
- 其次,真核生物中广泛存在可变剪切现象,使外显子和内含子的定位更为困难。因此,预测真核生物的基因结构需要运用更为复杂的算法,常用的有隐马尔科夫模型等。常用的软件有Genscan、SNAP、GeneMark、Twinscan等。
1.3.3 基因结构预测的研究内容
基因结构预测主要通过序列比对结合从头预测方法进行。
- 序列比对方法采用blat和pasa等比对方法,将基因组序列与外部数据进行比对,以找到可能的基因位置信息。常用的数据包括物种自身或其近缘物种的蛋白质序列、EST序列、全长cDNA序列、unigene序列等等。这种方法对数据的依赖性很高,并且在选择数据的同时要充分考虑到物种之间的亲缘关系和进化距离。
- 基因从头预测方法则是通过搜索基因组中的重要信号位点进行的。常用的软件有Genscan、SNAP、Augustus、Glimmer、GlimmerHMM等等。同时采用多种方法进行基因预测将产生众多结果,因此最后需要对结果进行整合以得到基因的一致性序列。常用软件有Glean,EVM等。
1.3.4 基因结构预测中拟解决的关键技术难点
目前,真核生物的基因结构预测方法仍有较大改进空间,主要面临以下的技术难点。
- 如何利用现有的数据和算法,更好地识别基因的可变性剪切位点。
- 随着测序工作的进展,许多目前研究较少的物种也将提上测序日程。大多基因结构的从头预测算法需要预先训练预测参数。现有资源和数据稀缺的物种将很难获得预测参数。
- 克服组装错误对基因结果预测的影响
1.3.5 建立基因结构预测的评价系统
可变性剪切位点的预测较为困难。如何结合RNA-seq数据进行可变剪切预测将是重要的工作方向和难点。
1.3.6 基因结构预测的研究方向
- 利用RNA-seq、EST等数据校正基因结构预测结果,识别可变剪切位点。
- 对于研究较少的物种,建议利用近缘物种的同源基因数据以训练基因结构预测软件。
- 利用同源基因组之间的共线性信息,辅助基因结构预测。
1.4 基因功能注释。
1.4.1 基因功能注释的研究背景和意义
获得基因结构信息后,我们希望能够进一步获得基因的功能信息。基因功能注释方向包括预测:
- 基因中的模序和结构域
- 蛋白质的功能
- 所在的生物学通路等
1.4.2 基因功能注释的发展现状
全基因组测序将产生大量数据,而实验方法由于成本较高,不适用于全基因组测序的后续功能分析。为此,目前普遍采用比对方法对全基因组测序的基因功能进行注释。KEGG和Gene Ontology是目前使用最为广泛的蛋白质功能数据库,分别对蛋白质的生物学通路和功能进行注释。Interpro通过整合多个记录蛋白质特征的数据库,根据蛋白质序列或结构中的特征对蛋白质进行分类。
1.4.3 基因功能注释的研究内容
目前,我们利用四个常用的数据库进行基因功能注释。使用的数据库有Uniprot蛋白质序列数据库、KEGG生物学通路数据库、Interpro蛋白质家族数据库和Gene Ontology基因功能注释数据库。
- 与Uniprot蛋白质序列数据库比对,获得序列的初步信息。
- 与KEGG数据库比对,预测蛋白质可能具有的生物学通路信息。
- 与Interpro数据库比对将获得蛋白质的保守性序列,模序和结构域等。
- 预测蛋白质的功能。Interpro进一步建立了与Gene Ontology的交互系统:Interpro2GO。该系统记录了每个蛋白质家族与Gene Ontology中的功能节点的对应关系,我们通过此系统便能预测蛋白质执行的生物学功能。
1.4.4 基因功能注释中拟解决的关键技术难点
目前我们的功能注释工作是建立在比对的基础上,这将会带来两个比较大的问题。
- 首先,此方法严重依赖于外部数据,对某些研究较少的物种限制很大。
- 其次,序列相似并不表示实际生物学功能相似,考虑引入序列比对之外的方法,进一步完善基因功能注释工作。
1.4.5 基因功能注释的研究方向
考虑引入序列比对之外的数据(如蛋白质互作网络、基因表达谱等),利用概率模型算法进行整合,完善基因功能注释工作。
二、自动注释所面临的挑战
随着测序技术的成熟以及测序成本的降低,对产生的越来越多的序列进行注释也显得尤为重要。微生物的基因组注释往往包括自动注释,以及之后的手动注释的矫正【2】.大多说的注释用的是同源性方法,通过与相近的已知的参考基因组比对,比对上,则认为有相近的功能。自动注释容易产生低质量的注释结果和错误,而手动注释则是为了去掉这些错误。但现在测序产生的大量数据,如果再用手动注释则显得不靠谱了。
高质量的注释不光仅仅是用一个软件预测一下ORF,然后获取跟他比对结果最好的已知的参考基因的信息。还应该包括ribosomal-binding sites (RBSs), termination sites and conserved motifs/domains.上面的这些注释不仅是为了更全面的注释一个基因,还可以更正之前注释产生的错误【29】。例如,通过RBSs和termination sites可以更群出知道基因真实的位置,这些软件有【4-8】.
如果仅仅依靠序列相似性来找注释有其局限性。我们获得一个新的序列,本来是想找它与其相近的序列的不同,却用相似性来注释它。而且如果参考基因组中没有相应的基因,也就可能漏掉我们感兴趣的区域。
随着数据量增加,如果仅凭注释出来的基因组的信息现在是很难上传到公共数据库中【9,10】。而现在二代测序中的RNA-seq则是结合实验结果,这样能够给相应蛋白的角色和功能有个更好的注释。这样的注释往往更可信,因为他不仅仅是根据序列相似性,还有实验的证明。目前,基因组的注释信息应该包含注释信息的的来源以及可靠性的依据,但是这一般被自动注释给省略了。包含证据信息可以给我们这注释信息的可靠性【11,12】.
1.细菌基因组的注释(不仅仅是比对注释)
- 在注释之前,一般用GLIMMER进行基因预测,这个软件是用参考序列来建立一个模型,然后用这个模型来预测我们感兴趣的基因组的基因【14,15,16】。
- 一旦找到orf,就用这些Orf去跟已知注释信息的参考基因组比对或者用比对工具(FASTA,BLAST)去跟整个UniProt数据库比对,得分最高则认为同源,则认为具有相似的信息。同时可以用其他的软件【20】来预测tRNAs,rRNAs.
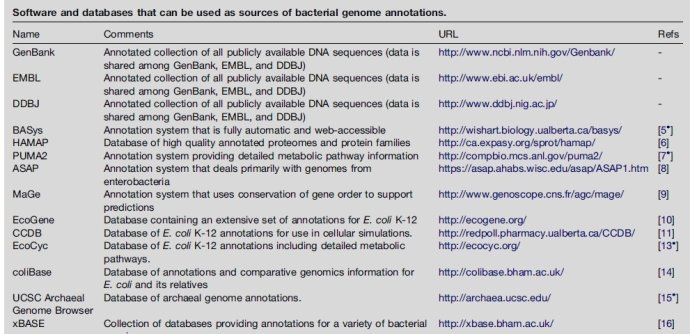
注释工具在之前那篇博文中提到了,网页版的有RAST [21], BASys [22], WeGAS[23] and MaGe/Microscope [24]; 本地化的有locally installed, such as AGeS [25], DIYA [26] and PIPA [27]. 还有MICheck [28]用来检查注释的语法错误。
基因预测软件有时给出错误的start/termination sites.通过找RBSs,我们可以推测出基因的start site,RBSFinder可以找到Shine-Dalgarno sequence模型。TransTerm用来搜寻rho-independent transcription来找termination sites。
在找到基因之后,蛋白的保守区域或模型应该添加到注释信息里面。有很多数据存储蛋白家族,例如ProSite, PRINTS and Pfam 【4, 7, 8】。InterproScan可以用来寻找区域或模型数据库(domain/motif databases)。 horizontal gene transfer (HGT)例如致病岛和噬菌体能够被预测出来,通过密码子组成和GC区别(在HGT区域和其余区域这两个会不同)【31】。他们也跟integrases, transposases and IS elements这些紧密相连【31】。有一些软件来预测他们【32,33,34】.
clustered regularly interspaced short palindromic repeats’ (CRISPRs) 和other tandem repeats是我们生物学家感兴趣的。例如,他们可以被用来发现细菌防卫机制和区别相近的菌株。软件(CRISPRFinder,CRT)和MICdb数据库(保存有预测的微卫星)可以用来预测他们。
蛋白的细胞定位能指示功能定位和发现药物靶标,有很多方法用来达到这个目的homology and keywords[40], amino acid composition [41–43] and a mixture of these [44], Gardy and Brinkman [45]写了一个这方面的综述。
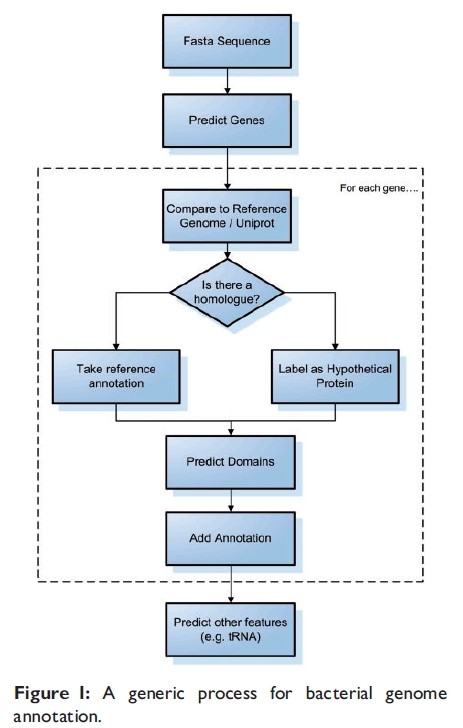
2.注释的局限性
如果注释较真起来会有很多的问题。你现在用来作为参考序列的数据也许是依赖更早的数据而得出的信息,错误的注释信息和错误也许能保留在新的基因组中,然后错误的信息最终带入到二代数据库中,例如UniProt,KEGGHE 区域特异性数据库PATRIC.公共序列数据库认识到了控制错误复制和提供可靠的软件来检查用户上传的数据的需要【9,10】。找到自动注释常见的一些错误,然后找方法来更正他们。
2.1注释的不一致性
同一个基因组用不同的工具注释出来的信息有时候也不同,常见的问题是分开/融合问题【48】.
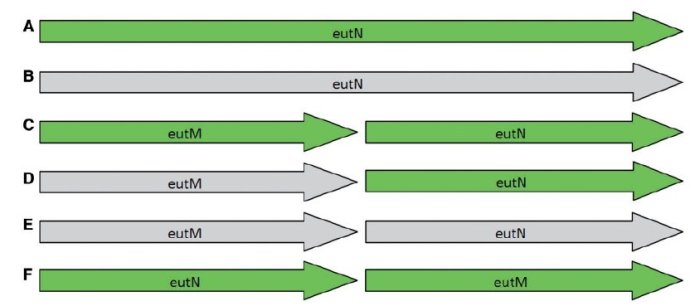
The six different models present across 17 RefSeq entries for Salmonella species for the eutM/eutN locus. Green indicates normal gene/CDS features, gray indicates gene features annotated as pseudogenes.(A) A single intact gene of 690 bp; (B) a single pseudogene of 690 bp; (C) two short intact genes 300 bp in length;(D) one pseudogene and one intact gene, each 300 bp in length; (E) two pseudogenes, each 300 bp in length; and (F) two intact genes with the order reversed.
说白了上图就是同一个基因在用不同的注释工具注释出来的结果不同,颜色浅代表是假设的注释结果。同名但是序列不一样。
直接预测区域(domains),而不是基因,用PfamAlyzer这个工具可能有助于找到区域中分开的基因。在上图的那个例子中,预测区域在所有的情况中会鉴别两个真实的区域,但是这个区域是来自一个还是来自两个基因则是没有办法弄清楚的。 最正确的注释是这个区域是把基因组跟这6个不同模型都比对一下,但是自动注释操作起来还是会有很多问题,如果结合实验数据(RNA-Seq)就更好了。
还有一种情况,序列同源但是不同名,如下图
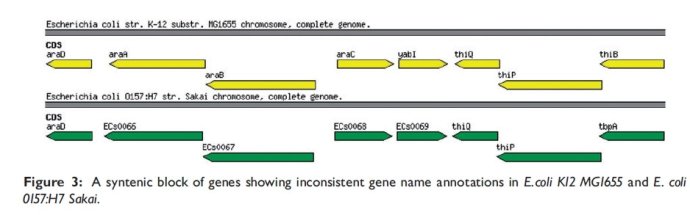
所以说嘛,参考序列选择的不一样,如果参考序列里面的注释信息不一致,那么注释出来的信息肯定也不一致,所以在选择相应的基因组来作为参考时,必须选择正确的包含最新信息的序列,同时未知的这个基因组不仅仅是跟一个参考基因组比较,跟其他的比较也是一样的。
2.2拼写错误
例如在UniProt中,有128个蛋白包含“syntase”,正确的应该是‘synthase‘.如果拿正确的拼写来搜索的话,会漏掉很多的信息。 手动来改掉拼写错误是一个巨大的工作量,能否利用一些能够提示错误的计算机语言【50,51】将会非常有帮助。
同一个基因名,不同产物名字
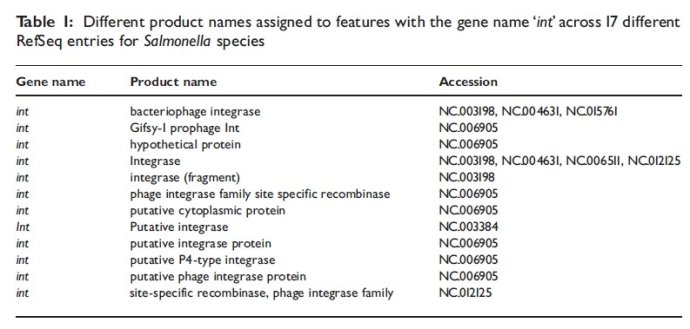
2.3假设蛋白
假设蛋白是那些被软件预测出来,但在数据库中找不到已知功能的同源基因,也找不到同源的已知功能域。他们可能是真实的未知功能的基因,也许是由于预测产生的错误。 许多细菌未知功能的基因根据他们在E. coli K-12的相对位置来命名为y-gene【52】 通常一些仅仅跟其他假设特征同源的特征,这些特征也灭有包含任何区域,现有的区域预测软件无法预测出来,是否注释这些特征也成为了一个思考的问题。正是基于这样,一些‘hypothetical proteins ‘被注释出来,发表和传播到新的数据库中,最后呈现在用户注释新的基因组的结果中。每个注释出来的特征的打分呈现出来也是非常有必要的,这样用户可以根据得分对可信度有一个初步的判断【12】。
对是否把这些蛋白放在注释信息里面有争议,如果他们的确是因为基因预测产生的错误,他们就应该被删除,否则他们会一直保存在第二代或第三代数据库中直到该蛋白被证实有功能。用Pfam和InterPro来寻找保守的区域或模型能让我们知道这个假设的蛋白是否有功能,但是这也有缺陷。事实上,蛋白有区域得分高并不是一定就说明有功能。Pfam[8], for example, contains over 3000 ‘domains of unknown function’, or DUFs, representing over 20% of known families [53]。
仅凭计算方法去得出一个基因组的区域是否有功能没有实际的意义,未知功能的保守特征也应该保留,这些特征可能会含有我们感兴趣的信息;但是他们应该跟我们有足够证明的其他的特征分开比较以让人知道他们的确信度是有不同的。
2.4直系同源和旁系同源(详见之前的博文)
同源意味着基因有一段共同的区段,直系同源来自基因的分化,旁系同源来自基因的复制。区分直系和旁系同源不仅对于进化关系至关重要,对功能关系也很重要,直系同源一般都保留相似的功能,而旁系同源随着时间的推移往往是具有不同的功能。
三、06年各种注释工具汇总
基因注释就是对测序出来的序列进行解释说明,把纯粹的碱基排列转化为有意思的生物学意义,告诉我们这个序列在生物体中起到什么作用。
注释的的核心就是预测序列的orf(也有人叫基因,也有人叫cds,用的工具比如说Glimmer),比对(跟已有信息进行比对,比如说blast,hmmer),数据库(根据已知信息的已知序列构建的可用来参考的数据库,比如说已有的数据库UniProt,Pfam,cog等)。
比对的思想则是原则相似的序列往往具有相似的功能。因此比对上了则认为两者具有一样的信息,而没比对上大体有三种原因:
- 序列与亲属有较大的差异;
- 代表的是一个未知的种属(之前没有发现过);
- 可能预测出来的Orf不对。
但是即使比对上了,就不是说一定具有相似的功能。同时数据库也不断在更新,而很多数据库越来越有针对性,比如针对微生物,或者针对微生物的某个属。用最新的数据库比对总是对的。
参考资料
- 放歌信天游 http://blog.sina.com.cn/s/blog_8698533a0101fxq5.html
- 丁香园 http://www.dxy.cn/bbs/topic/21530357 (感觉这个才是原创)
- 文献:The automatic annotation of bacterial genomes。(文中的编号来自该文献的编号)
- 文献:Automated bacterial genome analysis and annotation
