【3.3.4】Linclust序列聚类(基于Kmer,更快速)
Linclust已集成到免费GPLv3许可MMseqs2软件套件。 Linclust的源代码和二进制文件可以是在 https://github.com/soedinglab/mmseqs2 下载
随着高通量测序技术的迅猛发展,测序成本在不断降低,测序效率在不断提高。由此产生越来越多的生物序列数据,呈指数级爆炸增长,当然也包括基因序列或蛋白质序列。这些海量序列是后续生物信息分析的重要数据来源,一方面可以得到前所未有的生物信息,但另一方面也对数据分析研究者带来了巨大挑战,即数据的计算成本也在呈指数级增长。如何快速有效的分析海量序列数据是研究者面临的首要问题。
其中蛋白质序列聚类是数据分析的重要研究内容,蛋白质序列聚类一方面可以对序列进行去冗余操作,另一方面可以降低数据规模,进而方便数据存储和后续的分析步骤。因此许多算法研究者开发了许多蛋白质序列聚类算法,其中CD-HIT和UCLUST(USEARCH聚类版本)是目前可以处理海量蛋白质序列的两个经典聚类算法。目前这两个算法的谷歌学术引用次数(截止2019年3月27号)分别为4314(CD-HIT)和8912(UCLUST),是目前序列聚类领域的代表性算法,而UCLUST速度和影响力更胜于CD-HIT,有一种唯我独尊的地位。
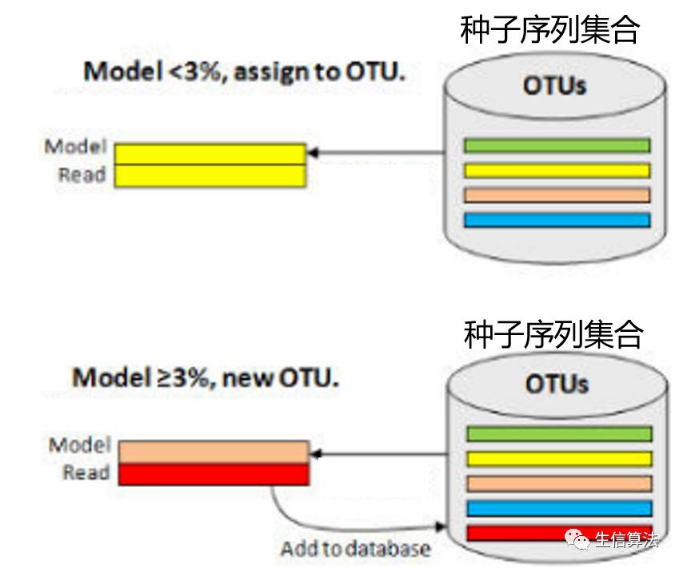
这两个算法都采用基于种子序列的启发式聚类思想,如下图所示。如果待比较序列和种子序列的距离(不相似性)小于给定阈值(如0.03),就将这条序列归入到种子序列所属类别。如果与所有种子序列的距离都大于给定阈值(如0.03),则将这条序列作为新的种子序列,加入到种子集合里。每条种子序列就是每个类的代表序列,由于每条待比对序列只与种子序列比较,避免了序列间两两之间的比较,大大降低了计算时间。所以CD-HIT和UCLUST的计算时间复杂度为O(KN),其中N 是序列的总条数,K 是种子序列的条数。
但在蛋白质序列聚类过程中,作者发现序列个数K 的大小与序列条数N 在同一个数量级,导致计算时间仍然很大,有时候甚至接近于N,因此时间复杂度有可能为O(N*N),并不是与N 线性增长。为了进一步降低运行时间,作者提出了Linclust算法,可将计算时间复杂度降低到O(N),比CD-HIT和UCLUST快上百倍,极大提高聚类效率。
UCLUST和CD-HIT不适用于序列差异性比较大的数据集
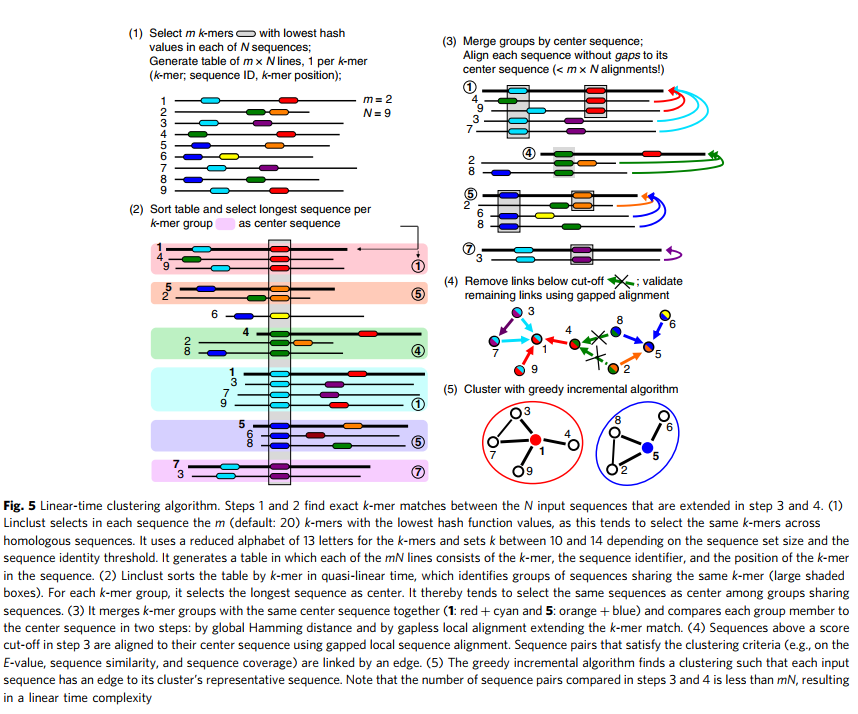
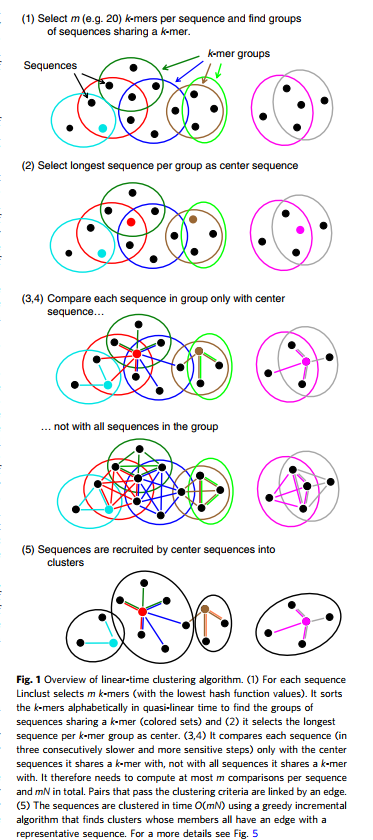
一、Linclust序列聚类算法步骤:
二、结果展示
3.1 运行速度比较
在UniProt蛋白质序列数据库进行比较,序列条数为61522444,比较的方法有CD-HIT、UCLUST等。下图是聚类时间的结果比较,相似性阈值分别设置为50%,70%和90%。可以看出Linclust方法在50%相似性阈值下,运行速度比UCLUST快2300倍,比CD-HIT快4600倍,比MMseqs方法快460倍,比MASH快69000倍,比DIAMOND快1600倍。在90%相似性阈值下,比CD-HIT快62倍,比UCLUST快100倍,MMseqs快570倍。

运行时间的指数级曲线,可以推断出Linclust运行时间与序列条数的关系是O(N 1.01次方),UCLUST是O(N 1.62次方),CD-HIT是O(N 2.75次方)。所以相对于其他方法,Linclust方法运行速度真正实现只与序列条数有关。
3.2 聚类灵敏度比较
聚类灵敏度(clustering sensitivity),作者定义为平均每个类的序列条数,平均序列条数越多,说明算法聚类灵敏度越好(a deeper clustering with more sequences per cluster implies a higher sensitivity to detect similar sequences)。下图是不同方法的聚类灵敏度比较,可以看出Linclust方法聚类灵敏度也不错。

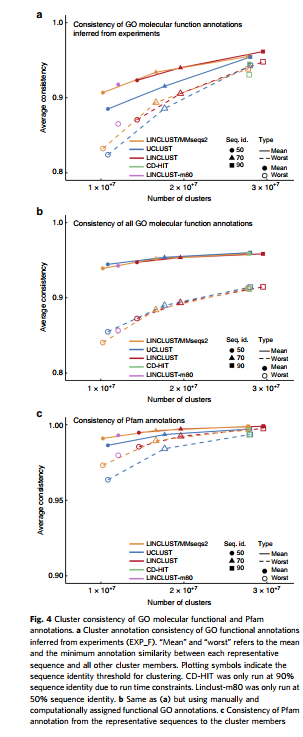
3.3 聚类一致性(consistency)比较
可以反映聚类的质量,用聚类结果的GO注释相似性分析,计算每个聚类单元里类成员与代表序列间的GO相似性,得出每个类的平均(mean)和最低(worse)的GO相似性。如下图所示,可以看出Linclust方法的聚类质量也不错,高于其他方法
参考资料
- https://blog.csdn.net/woodcorpse/article/details/106552935
- nature nature communications articles article. Published: 29 June 2018 . Clustering huge protein sequence sets in linear time. https://www.nature.com/articles/s41467-018-04964-5
