【7.1.2】由citizen科学家设计的de novo蛋白质
诸如GalaxyZoo,Eyewire和Phylo等在线公民科学项目已经证明在数据收集,注释和处理方面非常成功,但在很大程度上利用了人类模式识别技能而不是人类创造力。游戏EteRNA是一个例外,其中游戏玩家通过探索Watson-Crick碱基配对可能性的离散二维空间来学习构建新的RNA结构。然而,构建新蛋白质是一种在游戏中呈现的更具挑战性的任务,因为蛋白质结构的表示和评估本质上是三维的。我们在在线蛋白质折叠游戏Foldit中提出了从头蛋白质设计的挑战。向玩家呈现完全延伸的肽链,并且挑战制造折叠的蛋白质结构和编码该结构的氨基酸序列。在玩家设计的多次迭代,对得分最高的解决方案的分析以及随后的游戏改进之后,Foldit玩家现在可以从扩展的多肽链开始 - 生成多种蛋白质结构和在计算机中编码它们的序列。在合成基因中编码了具有与天然蛋白质无关的序列的一百四十六个Foldit player设计;发现56个表达并可溶于大肠杆菌,并在溶液中采用稳定的单体折叠结构。这些结构的多样性在从头蛋白质设计中是前所未有的,代表20种不同的折叠 - 包括在天然蛋白质中未观察到的新折叠。确定了四种设计的高分辨率结构,几乎与player型号相同。这项工作明确了大量隐含的知识,这些知识有助于从头蛋白质设计的成功,并表明公民科学家可以发现创造性的新解决方案,以应对蛋白质设计问题等突出的科学挑战。
一、前言
从头蛋白质设计的基本原理是蛋白质折叠到其最低的自由能状态;因此,设计一种新的蛋白质结构需要在规定的结构中找到具有最低能量状态的氨基酸序列。
在实践中,这个挑战可以分为两个子问题:
- 制定可设计的蛋白质骨架(即,可能是某些序列的最低能量状态);
- 找到制作结构中能量状态最低的序列。
蛋白质设计的挑战之一是多肽链可用的构象数量呈指数增长,即使对于60-100个残基的适度大小的蛋白质也是如此。因此,制作合理的骨架的第一个子问题是非常开放的,并且第二个子问题是困难的,因为明确检查设计的序列在制作的结构中具有比任何其他结构中更低的能量是不易处理的。近年来的从头蛋白质设计取得了相当大的进展,但尚不清楚是否所有这些成功的贡献都在用于设计蛋白质的方案中明确,以及隐含知识在专业知识中是多少。解决专业知识的作用尤其困难,因为第一个子问题(即,制定一个合理的骨架)所带来的极端开放式的挑战,实际上有无限数量的解决方案。因为不可能对骨架进行完整的计算机枚举,所以在生成和设计新蛋白质结构方面存在相当大的人类创造力和直觉空间。
为了研究基于群体的创造力如何有助于解决从头蛋白质设计问题,我们将de novo设计工具整合到蛋白质折叠游戏Foldit中。 Foldit是一款免费的在线计算机游戏,用于蛋白质建模中的众包问题,并提供对蛋白质模型的三维结构的完全控制(图1)。 玩家竞争建立具有最低自由能的模型,使用Rosetta能量函数计算。 在过去,Foldit主要应用于蛋白质结构预测问题,其中向参与者呈现非结构化氨基酸序列并且挑战以确定其天然构象。 在一个案例中,Foldit玩家重新设计了一个已折叠结构的环区域,但整个蛋白质的从头设计是一个更广泛的挑战。
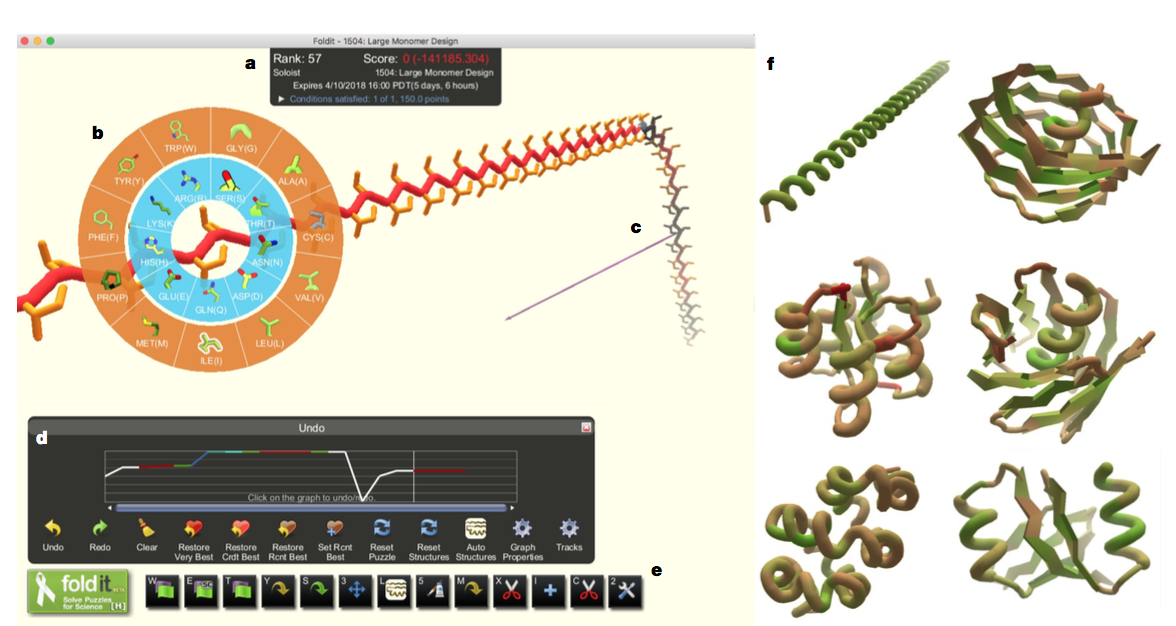
图1 | Foldit用户界面。 a,Foldit得分是具有负乘数的Rosetta能量,因此更好的模型产生更高的分数。 b,设计调色板允许玩家在模型的任何位置改变氨基酸残基的身份。 c,‘pull’工具允许玩家操纵模型的3D结构。 d,“撤消”图在跟踪模型开发时跟踪得分,并允许玩家回溯并加载模型的先前版本。 e,附加Foldit工具(从左到右):全结构最小化,侧链最小化,主干最小化,自动设计侧链,重新包装侧链,平移或旋转模型,二级结构分配,理想化二级结构,手动设计侧链,删除残基,插入残基,插入切点并理想化肽键几何结构。 f,Foldit玩家探索与天然蛋白质没有序列或结构同源性的多种结构。
我们反复挑战Foldit玩家从头开始设计稳定折叠的蛋白质,并根据他们的结果反复改进游戏。在每次挑战中,向玩家提供完全延伸构象(长度为60-100个残基)的聚异亮氨酸骨架,并给予7天将骨架折叠成紧凑结构并鉴定指定该骨架的序列。最初,大多数得分最高(低能量)的Foldit 玩家设计都是高度扩展的,缺乏溶剂难以接近的核心,完全由极性残留物组成(扩展数据图1);这种延伸的完全α-螺旋结构比折叠结构具有更有利的氢键,静电和局部扭转能量,这些结构必须扭曲以形成埋核。尽管聚赖氨酸和其他类似于这些初始Foldit溶液的扩展极性序列在溶液中通常是α-螺旋的,但缺乏长程相互作用阻碍了特定折叠成单一稳定结构。这突出了使用绝对能量作为蛋白质设计的优化标准的局限性:低能量设计并不能保证结构特异性,只有在所有其他替代构象具有更高能量时才会出现结构特异性。为了支持球状溶剂排除蛋白质折叠的设计,以及对它们进行独特编码的序列,我们在Foldit中引入了三个补充设计规则:
- 一个“核心存在”规则,需要最少比例的残留物(例如,30%)在设计结构中是溶剂可进入的;
- “二级结构设计”规则,禁止所有二级结构元素中的甘氨酸和丙氨酸;
- “残基相互作用能”规则,以惩罚在设计结构中造成分子内相互作用不足的大残基。
随着对Foldit的这些规则的增加,来自Foldit玩家的后续得分设计是紧凑的球状蛋白质。
扩展数据图1 |最初的最高排名的Foldit玩家设计。当挑战设计仅具有talaris2013评分功能的蛋白质(并且没有额外的规则)时,Foldit玩家发现了不太可能按设计折叠的低能量模型。 a,完全由赖氨酸和谷氨酸组成的延伸α-螺旋对氢键,静电和骨架扭转具有非常有利的能量,但不太可能协同折叠成单一的稳定结构。这种类型的设计不鼓励使用“核心存在”规则。 b,由于其较大的表面积,大的芳香族侧链可以比较小的脂肪族侧链产生更多的相互作用,即使在包装不足或溶剂暴露的情况下也是如此。这种类型的设计不鼓励使用“残留物相互作用能量”规则。 c,具有丙氨酸和甘氨酸饱和核心的设计可以在紧密堆积的骨架原子之间产生有利的范德华相互作用;然而,这些小侧链的埋藏与较弱的疏水效应相关,并且缺乏相互交叉允许具有相似核心堆积能量的多种构象之间的交换(即,熔球行为,molten globule behaviour)。这些设计不受“二级结构设计”规则的限制。
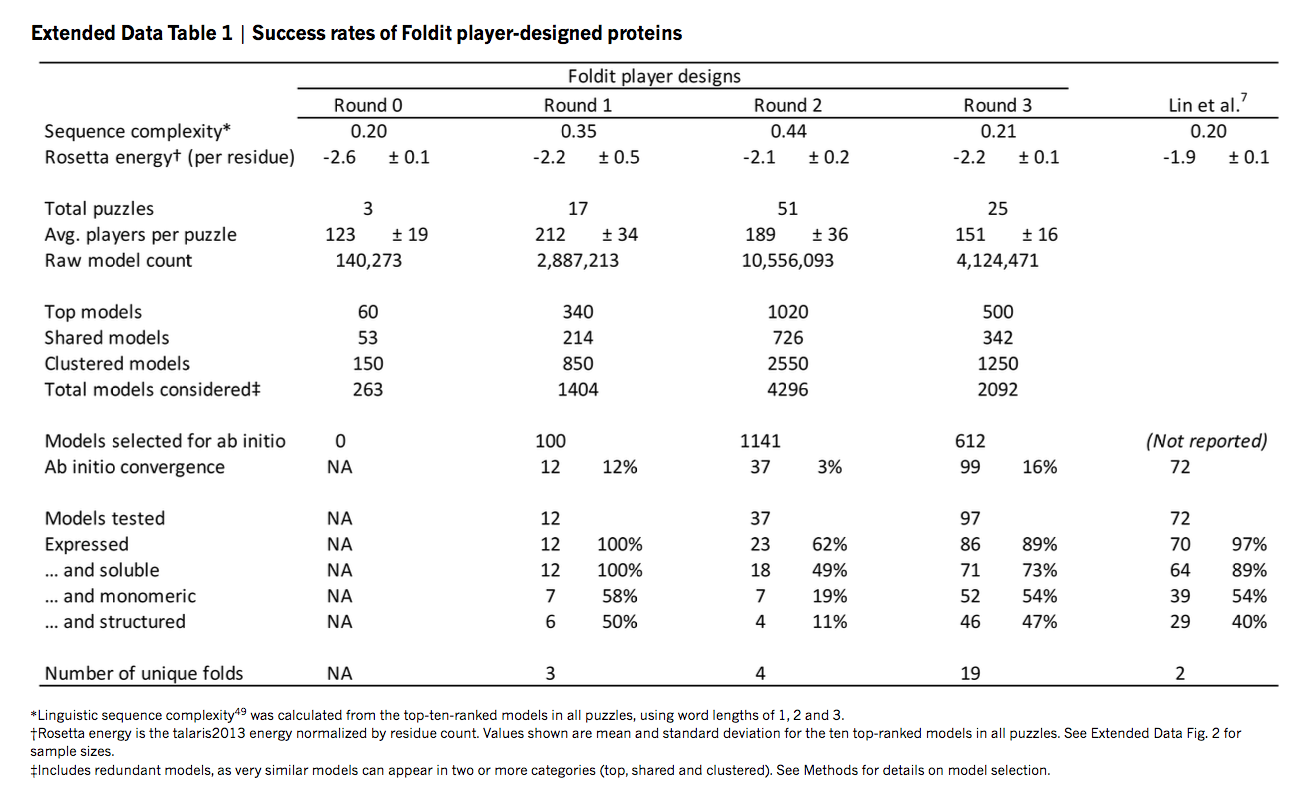
我们获得了编码12种玩家设计的定制合成基因,其结构预测计算包含在玩家设计的构象上。 这些蛋白质的序列与任何已知蛋白质没有同源性(补充表1)。 从头设计在大肠杆菌中表达,并通过金属亲和力和尺寸排阻色谱法纯化。 通过色谱和圆二色性分析表明,12种设计中的6种是单体的并且在溶液中折叠,螺旋二级结构与玩家模型一致(补充图1)。 本文中描述的所有经过实验测试的蛋白质完全是Foldit玩家的工作。
在游戏过程中,Foldit应用程序每2-5分钟将玩家的最新型号上传到Foldit服务器; 从这些快照中我们可以重建Foldit玩家开发蛋白质设计的过程(图2)。 与标准的Rosetta自动设计程序相比,Foldit玩家使用更多样化和复杂的探索策略,并且经常回复到他们模型的先前迭代以探索替代路径,从而产生高度分支的搜索树。 相比之下,典型的自动设计程序仅包括两个分支点。 此外,Foldit玩家定期采样比自动化程序更高的能量状态,自动化程序仅具有有限的能力来逃避局部能量最小值。

图2 | Foldit玩家和自动设计采样策略的比较。 a,三个Foldit玩家设计的红色(Foldit1),蓝色(Peak6)和绿色(Ferredog-Diesel)蛋白质的单一轨迹(忽略废弃的分支);并设计了四种Rosetta设计的灰色蛋白质的轨迹。 y轴是重新调整的Rosetta能量,因此最终设计的值为-1.00,正能量显示为零。 Foldit玩家愿意大量增加能量来探索新区域;相比之下,Rosetta程序具有有限的逃避局部能量最小值的能力。红色圆圈对应于b中所示的结构。 b,来自Foldit1设计轨迹的快照:(i)聚异亮氨酸的初始延伸链; (ii)二级结构的发展; (iii)折叠三级结构的发展; (iv)折叠结构的顺序设计,插图显示位置13和45处的有利包装; (v)高能中间体设计,插图显示在13和45位重新设计,导致与蛋白质骨架的空间冲突; (vi)最终精制设计,插图显示位置13和45处的重新有利的相互作用.c,Foldit1的设计策略表示为图形,显示了从单个中间体产生多个设计轨迹的所有分支点。最终设计是在17个分支点之后达成的。节点颜色对应于五个不同的合作Foldit玩家,最终设计标有星号。 d,Rosetta设计轨迹的类似表示 - 只有两个分支点。
受到Foldit公司从头开始设计稳定蛋白质的成功的鼓舞,我们对游戏进行了补充,以鼓励玩家探索更多样化的蛋白质结构。到目前为止,所有得分最高的Foldit设计都包含三个或四个通过最小环路连接的α-螺旋。实际上,Foldit玩家已经确定具有β-折叠的设计得分不如α-螺旋束(扩展数据图2),并且竞争性玩家放弃了任何设计更多变化折叠的尝试。通过实践科学家,这与蛋白质设计是一个有趣的平行,它也更多地关注螺旋束而不是其他类型的蛋白质折叠。为了鼓励设计更多种类的折叠,我们引入了“二级结构”规则,规定不超过50%的残基可能形成α-螺旋。 Foldit的玩家通过设计大量混合的α/β蛋白来应对,这些蛋白与目视检查的专家设计无法区分。然而,这些α/β设计序列的结构预测计算显示接近目标设计结构的不良采样,这表明设计的序列没有强烈编码其局部结构。进一步的分析表明,这些玩家设计包含许多具有局部应变骨架构造的残基(Ramachandran图的不利区域中的主干φ和ψ扭转)。这种设计具有非常低的能量,这表明当时的Rosetta能量函数存在问题:因为Rosetta用户通常从天然蛋白质的片段开始采样骨架,很少遇到不利的局部构象 - 因此,没有发现相关的能量当地骨架菌株被低估了。我们通过增加与应变局部骨架几何相关的能量惩罚的陡峭度来解决Rosetta模型中的这个缺陷;现在,这是在最新的Rosetta能量函数中实现的。我们还向Foldit添加了一个“理想循环”规则,该规则限制玩家使用19个非应变反向转换构想(unstrained reverse-turn conformations),并采用新工具来辅助生成无约束骨架:基于片段查找的循环闭合工具,交互式Ramachandran地图和用于二级结构元素和共同环构象的拖放组装的蛋白质蓝图方案(扩展数据图3)。这些升级共同带来了Foldit玩家设计蛋白质的局部骨架质量的显着改善(扩展数据图4)。

扩展数据图2 |排名最高的Foldit玩家的Rosetta能量设计。使用talaris2013评分函数计算顶级设计的Rosetta能量,并通过残基计数标准化。 a,排名前十的设计的能量来自:最初的Foldit谜题(第0轮; n = 30个设计),第1轮谜题(n = 170),第2轮谜题(n = 510)和第3轮谜题(n = 250) )。在第1轮和第2轮引入补充规则导致更高能量的设计(分别为P <10-6和P <0.01; Wilcoxon秩和检验)。第3轮中的主干建模改进导致了低能量设计(P <10-15; Wilcoxon秩和检验)。 b,使用“二级结构”规则(n = 220)从第三轮全α拼图(n = 30)或α/β拼图中排名前十的设计的能量。全α设计倾向于具有比α/β设计更低的能量(P <10-10; Wilcoxon秩和检验)。箱形图显示:中心线,中位数;箱限,上下四分位数;胡须,1.5×四分位数;点,异常值。
在实验表征中证实了降低局部骨架应变的重要性。 在前一段描述的骨架建模改进之前,测试的37个Foldit α/β设计中只有4个(11%)是单体的并且在溶液中结构化。 在骨架模型添加后,97个中的46个(47%)是单体的,并且在溶液中显示出预期的二级结构。 大多数在热和化学发现实验中表现出优异的稳定性,一些自由能展开(ΔGunf)超过20 kcal mol-1; 事实上,32种设计的蛋白质在95°C时完全折叠(图3,补充图1)。 这一成功率超过之前设计的α/β-蛋白质报告的成功率

图3 | Foldit玩家设计蛋白质的结构表征。 a,树状图显示所有56个折叠的Foldit玩家设计由结构相似性(TM-align)聚集,彩色圆圈突出显示以b-g表征的四种设计。堆叠条显示了聚类设计中的20个不同折叠(扩展数据图5)。折叠XX(见设计2003594_S028)是一种新的折叠,以前在天然蛋白质中未被观察到。 b-g,四种精选Foldit设计的卡通描绘(b); c-g中的图表对应于这四种结构。 c,Rosetta @ home ab initio计算表明,每个设计的序列都有一个强烈的能量景观,朝向设计结构。 y轴,Rosetta能量; x轴,设计的结构Cα rmsd;点表示从扩展链(红点)开始并从Foldit设计模型(绿点)开始采样的最低能量结构。 d,尺寸排阻色谱图(吸光度 在280nm处)表明设计在溶液中是单体的。 e,圆二色光谱表明,设计采用25°C(蓝色迹线)溶液中预期的二级结构含量,加热至95°C(红色迹线)并再次冷却至25°C(绿色迹线)。 f,圆二色性表示当温度从25℃升至95℃时残留椭圆率为220nm;设计不会随着温度的升高而变性。 g,用盐酸胍滴定过程中的协同解折叠。蓝色圆圈显示圆二色性意味着残留椭圆率在220nm随着变性剂浓度的增加而变化,黑色曲线显示出与数据拟合的双态展开模型。使用拟合模型参数27通过线性外推确定展开的自由能(ΔGunf)。
总体而言,56个成功的Foldit设计结构多样,代表20种不同的蛋白质折叠(图3,扩展数据图5),其中一个是以前在天然蛋白质中未观察到的新折叠。 Foldit设计的成功并不仅仅归功于一两个特殊的Foldit玩家,而是由Foldit社区广泛分享(补充表1)。 56个不同的Foldit玩家创造了56个成功的设计(最多产的玩家创造了10个成功的设计); 19个设计由至少2个合作参与者共同创建; 并且5个成功的设计并没有得分,但是被玩家标记为个人最爱。 Foldit玩家在蛋白质建模方面缺乏正式的专业知识(扩展数据图6,补充说明),但是在Foldit中玩蛋白质结构预测难题所获得的知识和直觉转化为从头蛋白质设计的成功(扩展数据图7)。
我们成功地解决了四种Foldit玩家设计的蛋白质的高分辨率结构。 三种设计蛋白质的X射线晶体结构(由其设计师Foldit1,Peak6和Ferredog-Diesel命名)与设计的构象紧密匹配,Cα均方根偏差(rmsd)分别为1.1,0.9和1.7(图4))。 Foldit1和Peak6的蛋白质核心中的良好分辨的电子密度表明大多数侧链采用预期的旋转异构体并保持设计的填充相互作用。 Ferredog-Diesel的电子密度不太清楚,但蛋白质主链采用设计的折叠,许多核心侧链似乎按预期包装。 第四种设 计Foldit3的溶液核磁共振(NMR)结构也与设计构象紧密匹配,具有Cα rmsd. 在设计模型和整体的medoid conformer之间的1.1Å。

图4 | Foldit玩家设计的蛋白质的高分辨率结构。 a,Foldit1设计(图3中的折叠V:三个β折叠,薄片顺序1-2-3)模型骨架(彩虹)与晶体结构(灰色)对齐,Cα rmsd 1.1Å。 b,Peak6设计(折叠III:四strands,sheet order 1-2-4-3)模型骨架对齐晶体结构,Cα rmsd 0.9Å。 c,Ferredog-Diesel设计(折叠I:四strands,sheet order 4-1-3-2)模型骨架对齐晶体结构,Cα rmsd 为1.7Å。 横截面显示核心残基侧链,复合省略2mFo- DFc图,轮廓为2.0 σ(a,b)或1.0σ(c)。 d,Foldit3设计模型(折叠XVII:四strands, sheet order 2-1-3-4)和NMR集合。 设计模型与具有的代表性(medoid)NMR模型对齐,Cα rmsd 1.1Å。 横截面比较设计模型中的核心侧链(彩虹)和代表性的NMR模型(灰色)。
从这些结果中,我们可以得出几个关于科学模型,公民科学和两者之间相互作用的一般性结论。首先,在实践科学家考虑的领域范围内的科学模型可能不属于这个领域。 Foldit玩家在他们的首次重新设计工作中产生的高度扩展的结构,以及后来由Rosetta用户先前未采样的局部几何形状的结构,可以最清楚地说明这一点。其次,对于公民科学家来说,通过在线游戏进行必要和创造性的科学贡献,游戏的评分功能必须是科学的准确表现。在我们最初的迭代中,Foldit并没有向玩家提供足够准确和通用的模型,以允许他们可靠地设计新的蛋白质,即使潜在的Rosetta软件已被实践科学家用于蛋白质设计。第三,也是最重要的是,公民科学通过模型试验和模型改进的迭代,提供了一种系统地改进科学模型的有效方法。人类游戏玩家非常有能力发现和利用未经验证的解决方案,这些解决方案是经验丰富的科学家无法探索的,他们的重点不是获得高分,而是解决他们的特定科学问题。
我们已经证明,玩在线电脑游戏Foldit的非专业公民科学家可以从头开始准确地设计全新的蛋白质结构。在本地,玩家的解决方案在物理上是合理的,类似于天然蛋白质,但在全球范围内,它们具有创造性和多样性。由公民科学家Foldit玩家设计的蛋白质绝不逊色于专业蛋白质设计者:它们准确地折叠到预期的构象,显示出优异的折叠稳定性并跨越多种多样的结构。鉴于几年前de novo蛋白质设计几乎完全没有解决问题,这一结果更令人印象深刻,成功的Foldit玩家模型所涵盖的蛋白质折叠的多样性大大超过以往任何蛋白质设计报告中的蛋白质折叠,我们的知识。 Foldit玩家对各种蛋白质折叠的持续成功突出了人类创造力的力量,在以易于理解的形式呈现的科学理解的指导下。
参考资料
- https://doi.org/10.1038/s41586-019-1274-4 ,David Baker,nature,2019 . De novo protein design by citizen scientists
