【4.7.1.4】MICAN--基于蛋白结构的比对
MICAN( a structurealignmentalgorithm that can handle Multiple-chain complexes, Inverse direction of secondary structures, Cα only models, Alternative alignments, and Non-sequential alignments )是由名古屋大学George Chikenji于2013发表在BMC Bioinformatics,一款蛋白结构比对的工具,它有如下特点:
- M) Multiple-chain complexes
- I) Inverse direction of SSEs
- C) Ca only models
- A) Alternative alignments
- N) Non-sequential alignments
设计该算法以通过忽略二级结构元件(secondary structure elements,SSE)之间的连接性来识别蛋白质对之间的最佳结构比对。 该算法的一个关键特征是利用每个SSE的多向量表示,这使我们能够正确地处理长SSE的弯曲或扭曲特性。
一、算法详解
假设我们有两个蛋白质结构,一个query(查询结构)和一个model(目标结构),分别由N和M个残基组成(后续,我们都以query和model来表示用于比较的两个蛋白结构)。 目标是找到两个结构之间最大程度相似结构的旋转矩阵,而忽略链的连通性。 MICAN的搜索方案采用分层对齐算法(hierarchical alignment algorithm ):
- 首先对齐SSE,
- 然后迭代地对齐残基。
SSE级别对齐进一步分为四个步骤:在第一步骤中,为两个结构的每个残差分配二级结构类型。在第二部分中,我们定义了“短段的比较元素(Comparing Elements of the Short Segment, CESS)”,通过它我们描述了在SSE级别对齐中要比较的结构。 CESS为连续3个strands残基,或者6个连续的helices。因此,每个SSE中包含的CESS的数量对于strands是n-2,对于螺旋而言是n-5,其中n是SSE的长度。每个CESS具有其SSE类型的信息,SSE段的代表性位置,从N末端到C末端的SSE的短段的方向,以及到SSE方向的垂直向量。因此,每个CESS都有两个向量,这使我们能够在每个CESS上定义一个参考框架。在第3步中,通过利用每个CESS拥有的两个向量,每个CESS定义一个参考框架。在第四步中,我们使用几何散列( geometric hashing )技术来获取最佳旋转矩阵的候选者,通过搜索CESS的最佳匹配来最大化非顺序结构的相似性。最佳旋转矩阵的一些候选者被列出并存储用于下一阶段。
残基水平比对分为两个步骤:在第一步中,我们基于通过SSE水平比对获得的叠加,进行残基水平比对。 从初始叠加开始,我们以Cα原子逐步形成一对一映射,其中我们从叠加较小RMSD的对的残基对对齐,逐步增加RMSD。在第二步中,迭代地执行对齐的细化,直到相似性得分收敛。 最后,基于TM-score like 函数选择最高得分的对齐方式。
(看完,还是不知道具体怎么回事,是不是?不急,每一步我们接着来看。。)
1.1 SSE级别对齐 The SSE level alignment
1.1.1 SSE标记
MICAN算法的第一步是标记query和model的所有残差的二级结构。 对于给定的残基,二级结构类型(α-螺旋,β-链或coil)用Zhang和Skolnick的方法标记,而不是DSSP或STRIDE,因为它的速度和仅适用于Cα模型。 据报道,相对于DSSP标记,该方法的Q3准确度为85%。 在SSE水平比对中,我们仅使用被指定为α-螺旋或β-链的残基,并忽略其余的残基。 我们还忽略了短于6个残基的螺旋和短于3的链。(这里定义了最小的比对单元)
1.1.2 定义短段的比较元素
在第二步中,对于query和model,我们定义了短段的比较元素(Comparing Elements of the Short Segment, CESS),通过它我们描述了在SSE级别对齐中要比较的结构。CESS为每个SSE的短段(Short Segment of SSE ,SSSSE),其中,SSSSE被定义为链的连续3个残基的strands,或的6个残基的helices。因此,每个SSE中包含的CESS( 或SSSSE)的数量对于strand是n-2,对于螺旋来说是n-5,其中n是SSE的长度(参见图12a和b)。
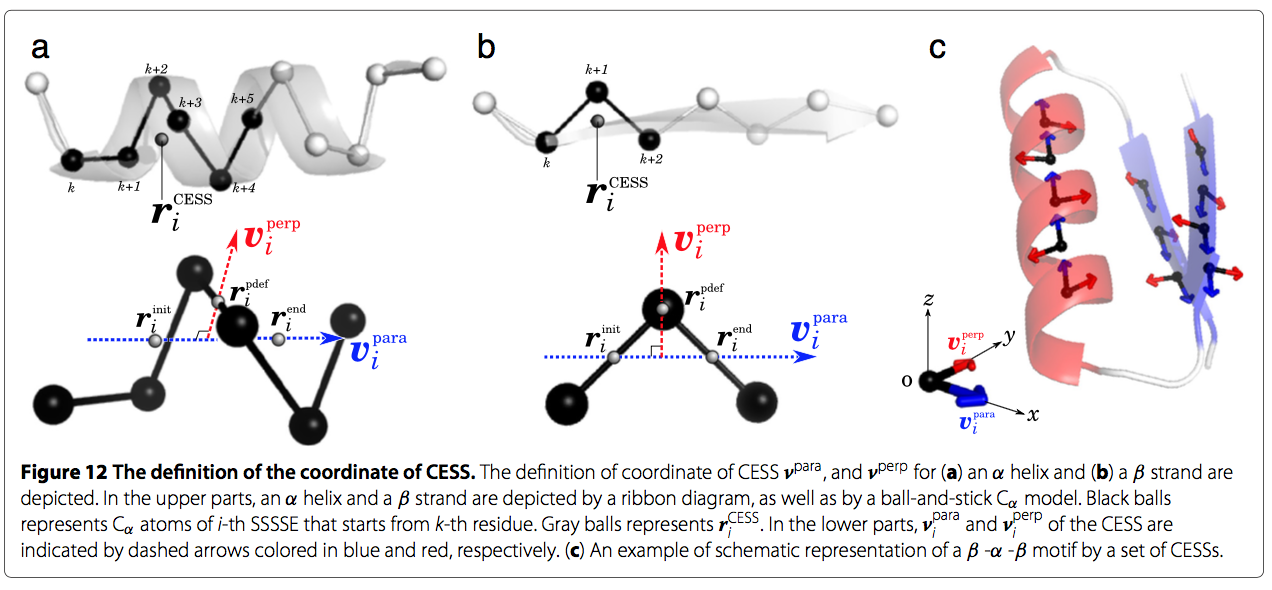
每个CESS都有以下四种关于SSSSE的信息:
1.第一个是在第一步中标记的二级结构类型(即α或β)。我们将第i个CESS的二级结构类型表示为$s_{i}$。
2.第二个是SSSSE代表点的坐标,我们称之为“CESS的坐标”。我们将第i个CESS的坐标表示为$r_{i}^{CESS}$。
当CESS表示从第k个残差开始的第i个SSSSE时(见图12a和b),$r_{i}^{CESS}$被定义为SSSSE两端的中间点,即
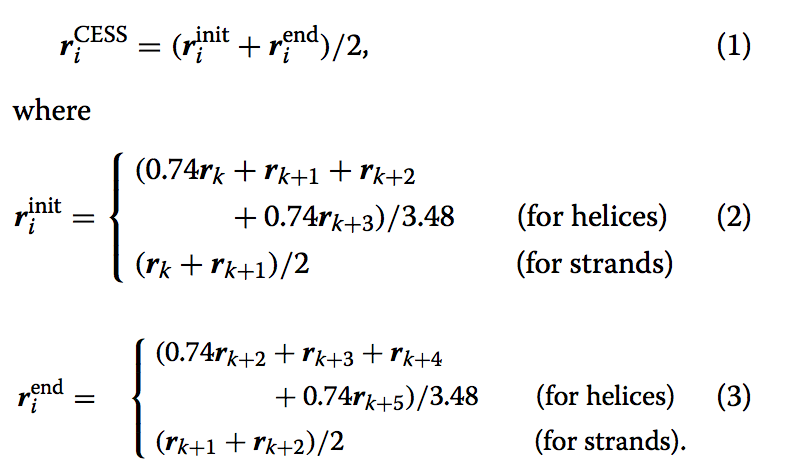
这里,$r_{k}$表示第k个残基的Cα原子的坐标。 $r_{init}$h和$r_{end}$表达式来自参考文献【14】。
3.第三个信息是从N-到C-末端的SSSSE的方向。第i个SSSSE的方向向量 $v_{i}^{para}$
$$ v_{i}^{para} = \frac{ r_{i}^{end} - v_{i}^{init}}{ | r_{i}^{end} - r_{i}^{init}|}$$
这个定义也参考文献【14】
4.第四个信息为$v_{i}^{para}$的垂直 向量 $v_{i}^{perp}$。我们引入这个向量的原因是通过引入向量我们可以定义每个CESS的参考框架。 这种技术使我们能够将几何散列算法的复杂度降低到O(n^2),而原始几何散列(其中每三个点定义参考框架)的复杂度为O(n^4)。(这里没有理解怎么把计算复杂度降低的!!!)。第i个SSSSE的垂直向量$v_{i}^{perp}$,为垂直于$v_{i}^{para}$,经过点 $r_{i}^{pdef}$。 $r_{i}^{pdef}$为SSSSE的中心点,
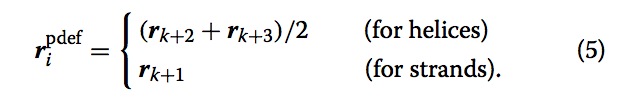
同样,我们计算每个SSSSE包含的这4个信息。 结果,query和model的整个结构由一组CESS描述。图12c中显示了一组CESS的蛋白质结构的示意图。
1.1.3 定义参考框架 defining the reference frame
在此步骤中,我们为query和model的每个CESS定义参考框架。 参考系是坐标系,用于几何散列技术。 如第二步所述,每个CESS具有两个彼此正交的矢量。 通过使用两个矢量,第i个CESS的坐标参考框架定义如下。 参考框架的原点为$r_{i}^{CESS}$。 x轴由矢量vi定义,y轴由vi定义。 z轴被定义为垂直于x-y平面,并且其方向由右手规则确定。(参考图12)
1.1.4 通过几何散列识别最佳旋转矩阵
为了找到最佳旋转矩阵的候选者,最大化两个结构的CESS的重合,我们使用几何散列技术。 几何散列算法由两个相互关联的阶段组成:预处理和识别。
在预处理阶段,我们为model结构设置哈希表。我们在这里使用的预处理过程基本上与标准过程相同,除了存储在哈希(hash)表中的一组描述。对于每个参考框架,我们定义3D网格。算法中网格的分辨率设置为3.2Å (为什么是这个数?)。网格的单元格构成哈希表,我们将其表示为H(a,b,c),其中(a,b,c)是3D立方体的索引。首先,选择model中的第i个CESS作为参考框架
的基础。其次,第j个CESS在参考框架i中的位置,我们称之为$r_{j}^{CESS}(i)$,根据公式计算: $r_{j}^{CESS}(i)≡ [(r_{j}^{CESS} -r_{i}^{CESS})·\hat{x}_{i},(r_{j}^{CESS} -r_{i}^{CESS})·\hat{y}_{i},(r_{j}^{CESS} -r_{i}^{CESS})·\hat{z}_{i}] $
,其中$\hat{x}_{i} $,$\hat{y}_{i} $,$\hat{z}_{i} $是参考框架的x,y和z轴的基矢量,$r_{i}^{CESS}$和$r_{j}^{CESS}$是i和j在原始坐标系中的坐标。
第三,如果在立方体(a,b,c)中找到$r_{j}^{CESS}(i)$,则第j个SSSSE的四个描述存储在哈希表H(a,b,c)中:(i)其SSE类型(sj)的标识,(ii)基础的标识(在这种情况下,定义参考框架i)的CESS,(iii)变换矢量$v_{j}^{para}(i)$,其是在参考框架i中计算的矢量$v_{j}^{para}$,(iv)变换矢量$v_{j}^{perp}(i)$。对所有参考框架的所有CESS重复该过程。
在识别阶段,我们比较query和model结构。 该步骤的目标是找到旋转矩阵的候选者,其最大化两个结构的CESS的重合。 除投票得分外,MICAN的识别阶段也与标准几何哈希算法基本相同。 令query结构的第k个CESS是定义参考框架的选定点。 对于query的每个其他CESS,计算由第k个CESS定义的框架系统中的坐标,model同样的操作。 这些坐标用作3D立方体的索引。 如果在立方体cube(a,b,c)中找到查询的$r_{l}^{CESS}(k)$),则model的框架系统i的第j个CESS的信息存储在哈希表H(a,b,c)中 ,给参考框架对i和k投票打分S(j(i),l(k))。 投票得分S(j(i),l(k))的函数定义为

这里,$ θ_{j(i),l(k)} $和$ φ_{j(i),l(k)} $分别是
$v_{j}^{para}(i)$和$v_{l}^{para}(k)$的夹角,$v_{j}^{perp}(i)$和$v_{l}^{perp}(k)$的夹角。对query结构的所有参考框架的所有其他CESS重复该过程。 结果,model和query之间的所有参考框架对根据它们的投票得分的总和进行排序。 最高得分的50个参考框架对被存储用于下一步(为什么选50个,后面会有介绍哦。。)。 对于每个这样的对,我们通过将一个参考框架叠加到另一个上来计算旋转矩阵。 这些旋转矩阵用于在残基水平对坐标矫正,建立两个结构的初始叠加。
1.2 残基水平比对
1.2.1 Cα原子的一对一分配
在该步骤中,从通过SSE水平比对获得的初始叠加开始,我们以逐步方式生成Cα原子的一对一映射,其中我们将来自与较小RMSD叠加的残基对对齐至较大。 为了识别哪个残差对与给定的截止距离dR叠加,我们基于query和model结构的叠合,构造N×M相似度矩阵$M_{i,j}(d_{R})$,其中N和 M表示query和model中的残基数。 矩阵$M_{i,j}(d_{R})$定义为

其中:
- dij是query的第i个残差的Cα原子与model的第j个残差的Cα原子之间的距离
- d0是标准化匹配差的标度
- dR是第R个步骤的截止距离
- σi表示残基i的二级结构(helix, strand or coil)的三态,δσiσj是Kronecker’s delta (这是一个二元函数,具体含义请百度)。
- 标度d0定义为
$d_{0} = 1.24 \sqrt[3]{L-15} -1.8$,其中L是query结构的长度。 该公式最初由Zhang&Skolnick引入,以消除评分函数固有的蛋白质大小依赖性[47]。 - 引入因子(δsisj+ w)/(1 + w)使得对齐的残基应属于相同的SSE类型。 这里,w是权重因子,在本文中设置为1.0。
- 截止值dR不同的的R的情况下对应的值为:d1 =3.2Å,d2 = 1.5×3.2Å,d3 = 2.5×3.2Å。
我们认为对齐仅由具有一定长度的连续段组成,以便消除可能是伪匹配的短段。 通常,长度为l的连续段被描述为一组残留对; {(α,β),(α+ 1,β+ 1),···,(α+ l-1,β+ l-1)},其中(α,β)表示query的残差α是 与model的残留β配对。 在这里,给定截止值dR,我们只考虑满足条件的连续段;
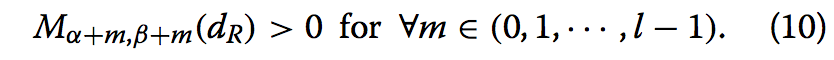
我们将这样的片段称为Aζ(dR),其中ζ是片段的索引。 我们引入了段Aζ(dR)的相似性得分S [Aζ(dR)],其被定义为
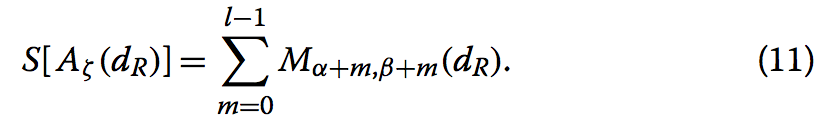
相似性分数用于对片段进行排名,以及排除短片段。
首先,我们将可重叠的残差对与最小截止值d1比较。 我们基于通过SSE水平对准获得的初始叠加来计算$M_{i,j}(d_{1})$,并基于该矩阵进行比对。 该步骤的目标是找到一组最大化的非重叠连续段{A1(d1),A2(d1),··,An(d1)}
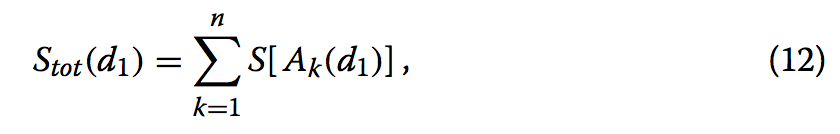
其中n是比对结果中的段数。由于很难获得这个问题的确切解决方案,我们的方法是使用类似贪婪的算法来找到近似解。首先,算法选择矩阵上所有连续段得分最高的连续段。我们将这样的段称为A1(d1)。如果S [A1(d1)]满足条件,则S [A1(d1)]≥Smin(哪来的Smin??),A1(d1)被记录为最佳段集合的第一个成员,其中设置截止参数Smin在后续中会提到。然后,为了选择下一个段,我们修改矩阵Mi,j(d1),使得干扰A1(d1)的矩阵元素被设置为零。换句话说,如果选择的段A1(d1)被描述为A1(d1)= {(α,β),(α+ 1,β+ 1),…,(α+ 1 - 1, β+ l - 1)},矩阵元素行为i(α≤i≤α+ l-1)或者其中列为j(β≤j≤β+ l-1)被设置为0,A1(d1)本身除外。我们还将段A1(d1)的矩阵元素设置为零,这样我们就不会在下面的步骤中选择A1(d1)作为最高得分段。接下来,我们选择在修改矩阵上具有所有连续段的最高得分的连续段。我们将这样的段称为A2(d1)。如果S [A2(d1)]≥Smin,则A2(d1)被记录为该组最佳段的第二个成员。此外,我们修改矩阵,使得干扰A2(d1)的矩阵元素以及A2(d1)的矩阵元素被设置为零。我们重复此过程,直到修改矩阵上所有剩余段的最高得分小于Smin。结果,我们获得了一组非重叠段,其近似最大化给定截止距离d1的总得分Stot(d1)。我们将这样的集合描述为{A1(d1),A2(d1),···,An(d1)(d1)},其中n(d1)是给定截止值d1的最佳段的数量。
其次,我们以逐步的方式将截止值dR从d2增加到d3来扩展比对。对于每个第R步,如在d1的情况下,我们根据等式(9)计算相似性矩阵$M_{i,j}(d_{R})$,其中N和。我们修改矩阵$M_{i,j}(d_{R})$,其中N和,使得干扰任何Ai(dR-1)的矩阵元素被设置为零,其中Ai(dR-1)是参与(R - 中获得的最佳对齐的片段)。 1) - 第一步。该操作确保在第(R-1)步骤中识别的最佳段的矩阵元素包括在第R步的最佳段中。然后,算法选择在修改的矩阵上具有所有分段的最高分数的分段。它被记录为第R步中最佳片段的第一个成员。我们将这样的片段表示为A1(dR)。接下来,我们进一步修改矩阵,使得干扰A1(dR)的矩阵元素以及A1(dR)的矩阵元素被设置为零。我们重复这些步骤,直到修改矩阵上所有剩余段的最高得分小于Smin,对于给定的截止dR。得到的最佳dR非重叠段集合描述为{A1(dR),A2(dR),···,$A_{n(dR)}(d_{R})$,},其中n(dR)是最佳段的数量对于给定的截止值dR。
通过重复从d2到d3的相同过程,我们最终得到d3的一组非重叠段,{A1(d3),A2(d3),···,An(d3)},用作初始值 迭代细化步骤中的对齐。
1.2.2 迭代细化
算法的最后一步是比对的细化(refinement of the align- ment)。 它如下迭代地执行。 首先,我们通过基于前一步骤中鉴定的比对残基最小化Cα原子的RMSD来叠加结构。 其次,基于叠加,我们通过残基水平比对的第一步中描述的方法获得新的比对。 重复这些程序,直到分数不再提高。 我们在这里使用的分数是修改后的TM分数,定义为
$$ mTM-socre = \frac{1}{N} \sum_{<i,j>}M_{ij}(d3)$$
N是query的长度,$\sum_{<i,j>}$表示对所有对齐的残基对进行求和。 最后,返回具有最高mTM分数的对齐。 尽管算法存在差异,但对齐通常会在中期收敛; 在叠合结构中,我们最小化RMSD而不是mTM得分,这是算法中的目标函数。 就我们测试而言,分数通常会收敛5-6次迭代,这表明差异并不重要。
1.3 选择最佳对齐方式
由于SSE水平比对中使用的评分函数与残基水平中的评分函数不同,因此在SSE水平比对中获得的最高评分叠加可能不总是导致残留水平中的最佳评分比对。我们探讨了两个评分函数之间的关系,发现虽然它们没有完美的线性关系,但两者之间存在很强的相关性。此外,我们确认如果我们从SSE水平比对的每个最高得分前50开始,我们在大多数情况下达到残基水平比对中的最高mTM得分。因此,为了获得尽可能高的残基水平比对的评分,MICAN从SSE水平比对的每个得分前50叠合结果开始,执行残基水平比对。在这50个比对中具有最高mTM得分的比对作为最佳比对结果返回。如果用户指定比对的输出选项,则MICAN程序可以输出次优比对结果。(这就是所谓的alternative)
1.4 处理反向比对 Handling reverse alignments
据报道,存在许多涉及反向比对的非顺序结构比对的有趣例子,其中SSE在结构上彼此匹配,但多肽链的方向相反。 如果需要,MICAN可以同时处理正向和反向对齐。 为了允许反向对齐,算法应更改如下:
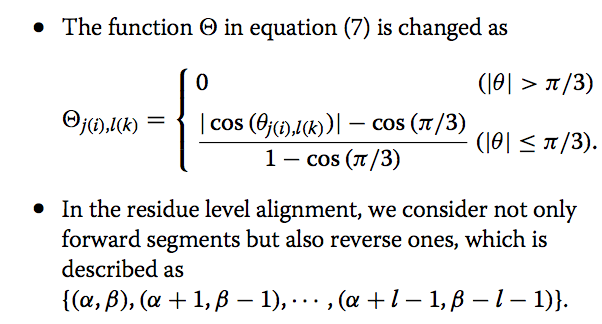
该算法在MICAN程序中实现。 用户可以选择仅forward或/forward/reverse混合模式。 本文中显示的所有数据均以仅forward模式获得。
1.5 参数调整 Parameter tuning
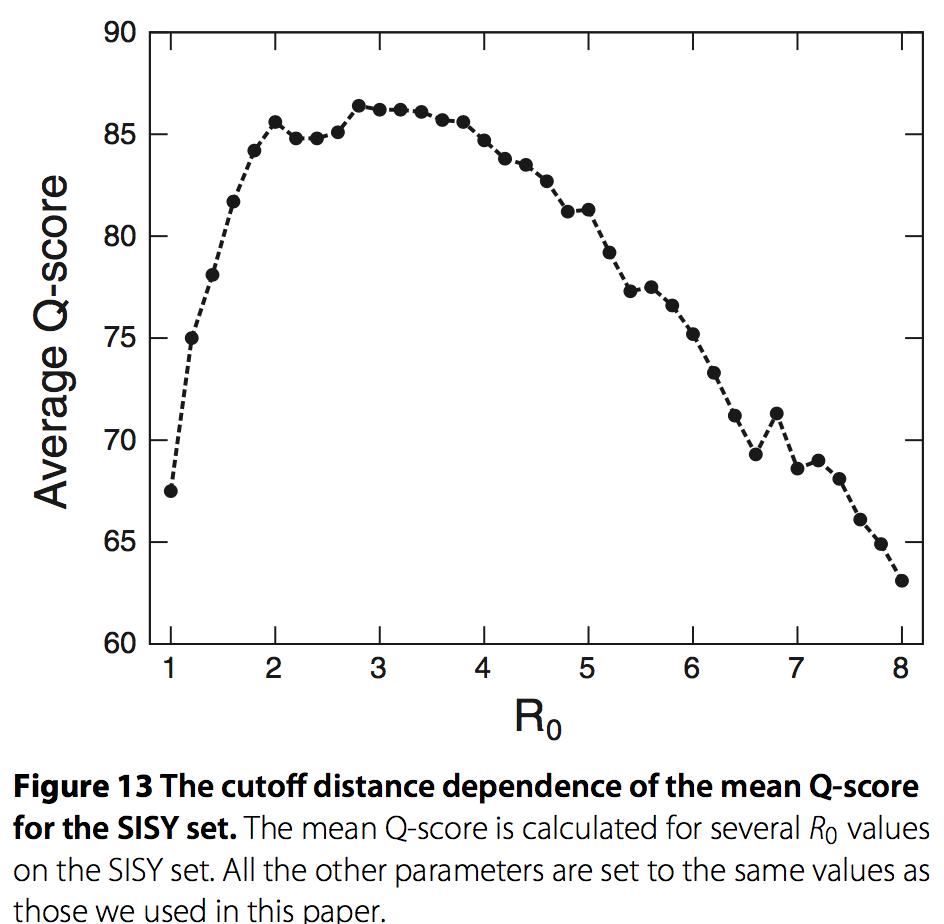
在MICAN算法中需要优化许多参数,例如方程中的w和dR。 我们通过非系统方式的try-and-error的方法,以最大化与SISY成对数据集的参考比对的一致性,来确定这些参数。 SISY-pairwise来自SISYPHUS数据库,SISYPHUS数据库是一个手动策划的多结构比对数据库,其包含具有非平凡(nontrivial relationships)关系的结构对齐,包括圆形置换或分段交换,因此该数据集适用于非顺序比对的训练。 通过训练,我们发现一些参数对性能有很大影响。 这些参数之一是在残基水平对准中使用的距离截止值dR。
在这项工作中,我们在每个步骤R中设置dR为d1 = R0,d2 = 1.5×R0和d3 = 2.5×R0,其中R0是截止参数。因此,单个参数R0确定dR的行为。图13显示了SISY测试集的平均Q评分和R0之间的关系。我们清楚地看到最佳值位于R0 = 3Å附近,并且当R0值从最佳点变化时,平均Q值得分急剧下降。另一个是几何散列算法中使用的体素大小(voxel size)h。我们观察到平均Q分数的体素大小依赖性与R0的情况定性相似。对各种dR和h值的初步数值试验,最后选择R0 = 3.2Å和h = 3.2Å。其他参数似乎对性能并不重要。训练结束后,MICAN最终与参考数据达成了高度一致(平均为86.2%)。该程序优于DaliLite(平均为77.4%),这是最好的结构比对软件之一。我们认为我们确定的参数集几乎是最优的,尽管尚未系统地优化。
1.6 数据集
1.6.1 顺序数据集 Sequential datasets
作为顺序测试集,我们选择MALIDUP和MALISAM,其包含具有non-trivial homology (MALIDUP)或结构类比(structural analogy)(MALISAM)的手动矫正的结构比对结果。 MALIDUP由241对蛋白质对和MALISAM 130对组成。 MALISAM测试数据和SISY训练装置之间没有重叠。 另一方面,MALIDUP测试集具有与训练集中使用的相同的三个蛋白质对。 但是,这三对仅构成MALIDUP测试集的1.2%。 因此,可以认为测试集几乎独立于训练集。
这两个测试集是最具挑战性的基准测试集之一,因为它们比其他数据库(如HOMSTRAD)包含更难的目标; 我们使用DaliLite进行了结构对齐基准测试,发现HOMSTRAD,MALIDUP和MALISAM的正确对齐残基对的分数平均值分别为90.1%,85.3%和67.3%。
1.6.2 非连续数据集 Non-sequential datasets
我们通过多段置换技术,基于顺序集MALIDUP-sq和MALISAM-sq创建了两个人工非顺序测试集“MALIDUP-ns”和“MALISAM-ns”。 该技术还用于生产合理的蛋白质结构诱饵组( reasonable decoy sets of protein structures ),保留了原子的自然空间排列。 创建方案描述如下(也参见图14):
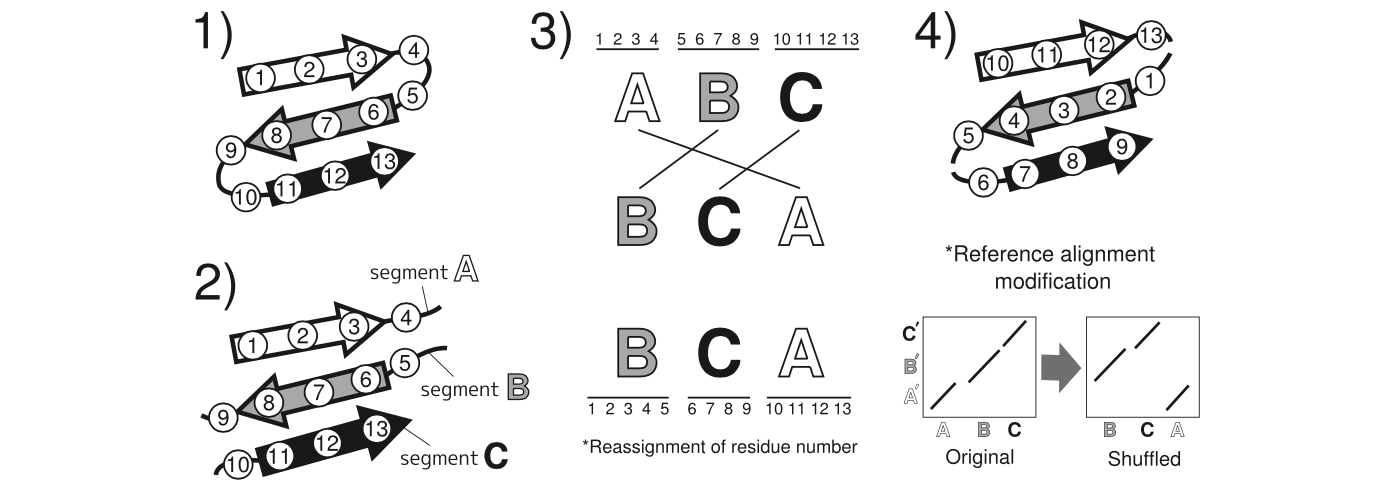
图14 如何创建非顺序测试集的示意图。(1)从每个靶对中随机选择一种待置换的蛋白质结构。原始残留数字显示在圆圈内。三个SSE(股)用箭头表示。 (2)原始链在环路(loop)位置被分成几个部分。在该示例中,链被分成三个段A,B和C.(3)段顺序被随机混洗(例如,顺序A-B-C被混洗成不同的顺序B-C-A)。根据混洗的分段顺序重新分配残基数。 (4)通过置换技术生成的结构。其残基数也根据分段顺序进行修改。根据置换蛋白的重新分配的残基编号修饰参考比对。 A',B’和C’代表另一种蛋白质结构的片段,其未被选择用于序列改组。段A',B’和C’分别在结构上对应于段A,B和C.
- 我们从每个目标对中随机选择一个蛋白质结构,并通过DSSP程序识别环区域(loop regions)。
- 在所有环区域将链分成几个区段。
- 随机改组这些段的线性顺序,并相应地重新分配残基数。 我们没有为那些置换结构执行循环建模。
- 根据重新分配的残基数修改每个目标对的参考比对。
通过这种方式,我们从每个顺序参考对齐生成了一个非顺序参考对齐。 对所有顺序测试集执行相同的过程,产生来自MALIDUP-sq和MALISAM-sq的241和130个非顺序对齐。 我们在此处使用的顺序/非顺序测试集可在 http://www.tbp.cse.nagoya-u.ac.jp/MICAN 在线获得。
二、背景介绍
蛋白质结构比对技术,对于推断不能仅从序列相似性中推断的比较远的进化关系,并且在蛋白质中发现重复的结构基序,有重要的作用。由于其重要性,已经开发了许多结构比对算法。尽管存在多种结构对齐算法,但是它们中的大多数遵循简单的顺序规则,即,两个蛋白质仅按顺序排列。然而,最近报道了许多显示非顺序结构相似性的有趣实例,其中非连续结构相似性是结构相似性,其中结构等同区域在比较蛋白质的序列中以不同顺序排列。出现最多的非顺序结构相似性是循环排列( circular permutation )。尽管没有循环排列的那么多,但是显示出超出循环排列的非顺序相似性的示例的数量也被证明是不少的。此外,Ilyin等人发现蛋白质之间的非顺序结构比对不限于任何特定折叠的蛋白质,并且它们在整个蛋白质世界中是系统的和广泛的,表明非连续结构关系的通用性和重要性。
非顺序结构相似性在进化方面是有趣的。目前,已知具有进化关系证据的蛋白质对的一些实例,其折叠成拓扑上不同但共享相同的二级结构填充排列。这种蛋白质对的最有趣的例子之一是hnRNP K( PDB entry 1KHM)和核糖体蛋白S3(PDB entry 1J5E)的KH结构域。它们显示出统计学上显着的序列相似性(38%序列同一性),暗示同源性。然而,它们的结构在拓扑结构上完全不同,同时融合到相同的架构(architecture)中。另一个例子是一对羧酸酯酶(PDB entry 1YAS)和Hydroxynitrile lyase(PDB entry 1QLW)。两者都属于SCOP数据库[12]中定义的相同超家族(α/β水解酶超家族),表明它们共有一个共同的进化祖先。然而,结构对齐揭示了长结构等同区域的序列顺序被交换。这些实例表明,一些蛋白质通过片段重组或重排事件改变了它们的折叠,同时保留了相同的核心包装排列。对这些蛋白质对的进一步研究将为我们提供关于蛋白质如何在进化过程中改变其折叠的暗示。
尽管已经开发了一些非顺序结构比对方法[13-22],但非顺序结构对齐算法仍有很大的改进空间。实际上,我们已经测试了所有公开可用的非顺序结构比对程序,并且发现了当前存在的方法的一些问题,这些问题是非顺序结构比对特有的问题:
- 问题之一是利用每个SSE的单个矢量表示的一些非顺序结构比对算法,倾向于在对齐长SSE的残基时失败。这种简化的蛋白质表示对于非顺序比对尤为重要,因为非顺序比对的搜索空间远大于顺序比对的搜索空间。然而,这种方法不适合描述长SSE的弯曲或扭曲性质,这使得难以正确地对准长SSE区域。而且,即使长SSE没有弯曲或扭曲,也可能难以通过单矢量表示正确地对准长SSE中的短SSE。
- 另一方面,采用蛋白质结构的Cα表示的另一种非顺序结构比对方法具有另一种问题。例如,SAMO产生的比对,试图最大化匹配的Cα原子的数量并最小化它们的均方根距离,经常显示过度碎裂;尽管匹配残基的数量很大并且排列的残基的RMSD很小,但没有长的连续排列区域。对于芳胺N-乙酰转移酶和附睾视黄酸结合蛋白,图1中显示了这种比对的实例。这两种结构非常相似,如参考对齐所示,它来自MALISAM数据库[23],这是一个结构相似的对的手动矫正的数据库。对于相同的蛋白质对,SAMO产生完全不同的比对。参考比对具有超过77个残基的4.4 Å 的RMSD,而SAMO的比对在91个残基上给出3.4 Å+,表明SAMO的比对仅比这些值更好。然而,这种高度分散和分散的对齐是否具有进化性或物理意义是值得怀疑的。
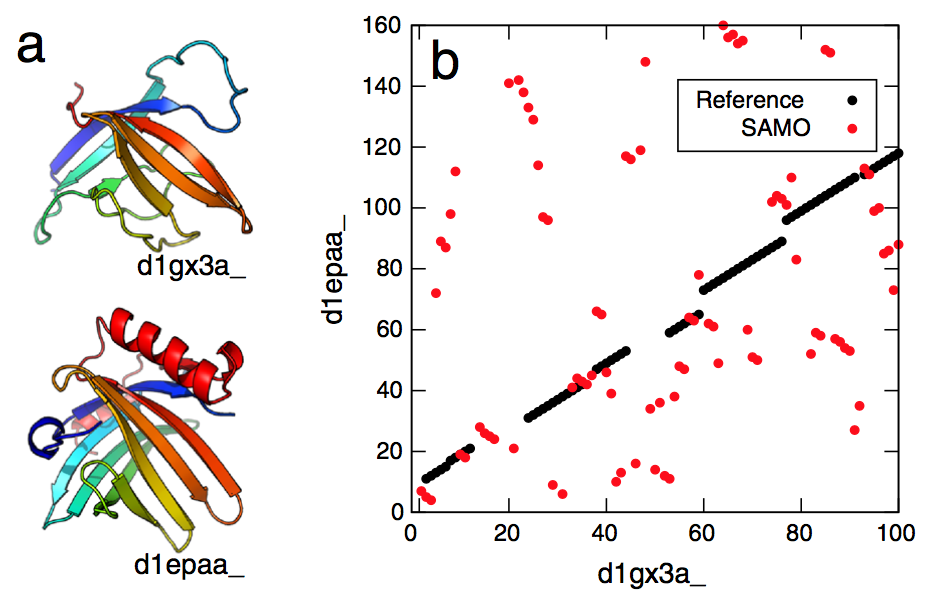
图1 对齐中“过度碎片化”(over-fragmentation)的说明性示例。 通过非顺序结构对齐程序SAMO将手动矫正的结构对齐与自动生成的对齐进行比较。 (a)芳胺N-乙酰转移酶(SCOP-ID d1gx3a,上部显示)和附睾视黄酸结合蛋白(SCOP-ID d1epaa,如下部所示)的结构。 这一对来自MALISAM数据库。 (b)显示该对的对齐图。 黑点表示参考对齐,红色表示SAMO对齐。 参考比对显示在77个残基上的RMSD为4.4 Å,而SAMO的比对在91个残基上产生3.4 A +。
在本文中,我们介绍了一种新的非顺序结构对齐方法,称为“MICAN”,克服了上述问题。设计MICAN是为了找到最大化SSE区域结构相似性得分的旋转矩阵,而不管链条连通性如何。由于MICAN在结构比较中使用SSE作为结构单元,因此它没有过度碎裂问题,或者没有主要由相对短的区段或单个残基组成的噪声排列。 MICAN的搜索方案基于几何散列。它最初是为计算机视觉领域的模型的物体识别问题而开发的,并且已被广泛应用于蛋白质结构比较研究[13,24-27]。几何散列具有不使用要比较的点的顺序信息。因此,在忽略顺序的情况下,它非常适合于结构比较。该算法的关键特征之一是利用每个SSE的多向量表示。此功能使我们能够正确处理弯曲或扭曲的长SSE。另一个特征是几何散列技术中的参考框架构建方式,其中每个要定义的点定义一个参考框架,而不是每对或每个三元组定义。借助于这种技术,我们可以减少要与O(n)进行比较的参考框架系统的数量,而每个三元组定义一个参考框架的情况的数量是O(n^3),其中n是要比较的要点的数量。因此,MICAN算法非常快;它是我们在这里比较的所有程序中最快的非顺序结构对齐程序。
为了评估算法的性能,我们将MICAN与其他公开可用的非顺序对齐程序进行比较:DEDAL,SNAP,GANGSTA +,SCALI,MASS和SAMO,以及顺序的:DaliLite,CE和TM-align。 我们在顺序和非顺序基准测试集上,使用依赖参考( reference )和不依赖参考评估方法评估这些程序。 我们证明MICAN优于其他现有方法,用于再现非顺序测试集的参考比对。 此外,虽然MICAN并不专注于顺序结构对齐,但它显示了在依赖于参考和不依赖参考的顺序测试集中的顶级性能。
三、结果与讨论
3.1 评估
为了尽可能客观地评估比对方法的性能,我们使用参考依赖性和参考独立评估方法。
依赖于参考(reference )的评估基于参考比对预先计算的金标准。 通常仔细检查参考对准,或手动矫正以确保良好的对准质量。 它们被认为在大多数情况下具有生物学或物理意义,尽管它们可能不一定是完美的。 参考依赖性对准精度计算,为测试比对中正确比对的残基对的数量除以参考比对中的比对残基对的总数。 由于其生物学或物理相关性以及评估的简单性,已经广泛使用依赖于参考的评估。 但是,该方法的某些方面受到了批评; 它在很大程度上依赖于参考比对的正确性,并且不清楚参考比对的质量如何影响比对程序的评估。
另一方面,reference-independent 的方法不需要参考比对。它评估纯粹的几何测量,例如对齐的残基对的数量,它们的RMSD,间隙数或它们的组合。因此,与参考无关的方法没有与依赖于参考的评估相关的问题。比较这些几何测量对于识别几个对准程序的特征特别有用。然而,这些相似性度量可能不一定反映生物学或物理上有意义的相似性[34]。这种情况特别严重,特别是对于非顺序结构对齐。例如,如图1所示,SAMO的比对显示出比reference alignment更大数量的比对残基对和更低的RMSD。然而,它的对齐似乎在物理上并不相关,因为在对齐中有太多的间隙和太多的短段可能是虚假的匹配。 如上所述,参考依赖和参考独立方法都有其自身的优点和缺点。两种评估方法的使用将为基准测试的结果提供更客观的理解,突出显示算法的不同方面。
3.2 数据集
我们应该使用哪种数据集来评估非顺序结构比对程序?
显然,需要许多“正确”比对且非连续的蛋白质对。 我们将这些测试集称为“非顺序测试集” ( non-sequential test sets ) 。除此之外,还需要“顺序测试集” ( sequential test sets ),即“正确”比对顺序的蛋白质对的集合。 这是因为理想情况下,如果正确对齐是顺序的,则非顺序比对程序应该也能生成顺序对齐。 顺序测试集使我们能够评估非顺序程序的这种能力。 因此,我们使用顺序和非顺序测试集来评估不同的结构比对程序。
作为顺序测试集,我们选择MALIDUP和MALISAM,其包含具有非平凡( non-trivial )同源性(MALIDUP)或结构类比(MALISAM)的手动矫正的结构比对。 我们将这些基准集称为“MALIDUP-sq”和“MALISAM-sq”。
由于缺乏非顺序结构比对数据库的黄金标准,我们必须构建用于评估非顺序比对的非顺序测试集。 在这里,我们基于顺序测试集创建人工非顺序测试集“MALIDUP-ns”和“MALISAM-ns”,而不是收集自然发生的非顺序比对,因为当前已知的天然存在的蛋白质对的数量显示 非顺序结构相似性是有限的。 有关如何创建人工非顺序测试集的详细说明,请参阅“方法”部分。
3.3 与其他比对软件的比较
我们将MICAN的比对与其他结构比对程序的比对进行了比较。 它们是三个顺序比对程序:DaliLite,TM-align和CE,以及六个非顺序比对程序:DEDAL [33],SNAP [22],GANGSTA + [21],MASS [13],SCALI [16]和 SAMO [18]。 我们选择DaliLite,TM-align和CE作为顺序比对程序的代表,因为它们已被认为是最好的结构比对程序之一[4,6,29]。 上述非顺序程序的选择基于独立程序的可用性。 所有程序都在Linux平台上本地下载和执行。 如果该程序的源代码可用,我们在计算机上使用GNU Compiler 4.5.5编译它。 据我们所知,这是目前对非顺序结构比对程序进行的最大和最全面的比较。
3.4 reference-dependent 评估的基准测试
3.4.1 顺序比对测试的结果
为了评估几种方法在顺序测试集上的比对精度,我们将10种方法生成的比对与MALIDUP-sq和MALISAM-sq的所有参考比对进行了比较,并计算了一致性百分比,Q-score。这里,Q-score被定义为测试比对中正确排列的残基对的数量除以参考比对中的比对残基对的总数。相应的箱线图如图2所示,平均Q-score列于表1。我们还给出了图3中所有对比方法的Q-score散点图。对于MALIDUP-sq测试集,MICAN获得的最大平均Q评分为85.8%(中位数为94.9%),DaliLite为第二大平均值为85.3%(中位数为94.3%)。另一方面,对于MALISAM-sq测试集,最好的算法是DaliLite,第二个最好的是MICAN。 DaliLite的平均Q评分为67.3%(中位数为80.7%),MICAN为65.5%(中位数为85.9%)。 DaliLite和MICAN的比对精度在统计上无法区分;根据Wilcoxon signed rank test,MALIDUP-sq和MALISAM-sq的p值分别为0.602和0.937。这两个程序是比对中的主要方法,因为这两个程序之间的差异具有统计学意义,P <0.01。例如,MICAN和DEDAL的比对精度在统计上是可区分的; MALIDUP-sq和MALISAM-sq的p值分别为9.7×10-5和2.7×10-3。图3中显示的Q-score的散点图也显示了两个程序的优越性。从这些结果可以得出结论,MICAN和DaliLite至少对于MALIDUP-sq / MALISAM-sq测试集的reference-dependent评估显示出最佳性能。应该注意的是,尽管MICAN并不专注于顺序结构比对,但MICAN的性能与DaliLite的性能相当,DaliLite已知是最佳顺序比对程序之一。

图2使用顺序测试集的参考依赖评估结果。 对于三个连续(白色框)和七个非连续方法(灰色框)获得的Q分数分布的箱线图。 左(右)图表示使用MALIDUP-sq(MALISAM-sq)设置的测试结果。
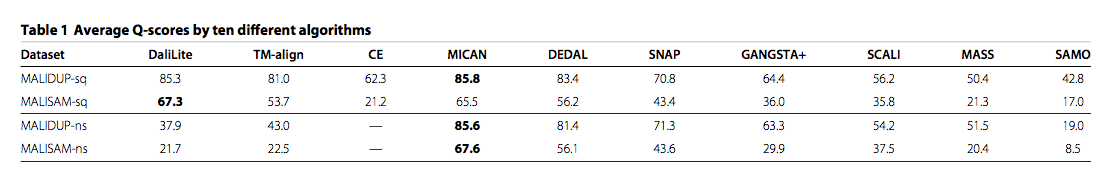

图3 连续测试集的所有成对比较方法的Q分数散点图。 比较顺序测试集的所有成对比对程序的Q分数。 右上角对角线显示MALIDUP-sq集的散点图。 左下角显示MALISAM-sq集。(每个点的横纵坐标即为不同方法得到的Q分数)
3.4.2 非顺序比对测试
为了检查非顺序参考比对的能力,我们对非顺序测试集,MALIDUP-ns和MALISAM-ns测试集进行了基准测试。就我们测试而言,CE程序不适用于非顺序测试集中的蛋白质对。这是因为测试集中包含的人工产生的蛋白质结构具有链断裂。因此,没有针对测试集显示CE算法的数据。非顺序基准测试的结果显示在图4,图5和表1中。正如我们所预期的,顺序对齐程序DaliLite和TM-align,与其在顺序测试集上的结果进行比较,非顺序性能上显着降低。例如,DaliLite获得的平均Q分数从85.3%(MALIDUP-sq测试)下降到37.9%(MALIDUP测试)。该结果是自然结果,因为这些顺序比对程序未设计成以非顺序方式比对蛋白质结构。另一方面,非顺序对齐程序的性能对于从顺序到非顺序的测试集的变化是稳健的。其中,MICAN在测试装置上表现出色。它在非顺序测试集上实现了最高的中位数和平均Q评分(参见图4,图5和表1)。特别是,MALISAM-ns测试的结果是更难的,这是更重要的; MICAN获得的平均Q评分为67.6%,比DEDAL高出11%,显示第二高的平均Q评分(见表1)。此外,MICAN和DEDAL之间的差异具有统计学意义:p值为1.7×10-4(另见图4)。结合顺序和非顺序测试集的结果,无论参考对齐是否顺序,至少对于我们使用的测试集,MICAN可被视为用于生成与参考对齐一致的比对的优秀程序。
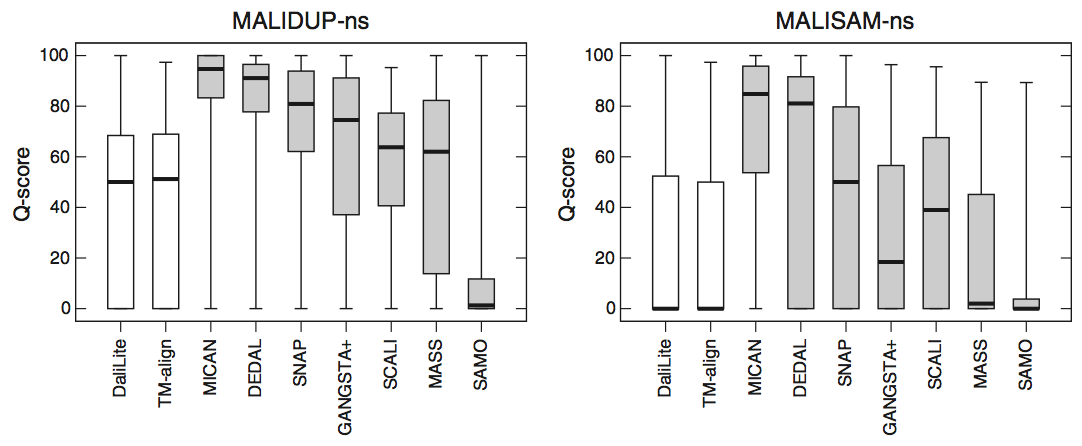
图4 使用非顺序测试集的参考依赖(reference-dependent)评估结果。 对于三个连续(白色框)和七个非连续方法(灰色框)获得的Q分数分布的箱线图。 左(右)图表示使用MALIDUP-ns(MALISAM-ns)设置的测试结果。

图5 非顺序测试集的所有成对比较方法的Q分数散点图。 比较非顺序测试集的所有成对比较方法的Q分数。 右上角显示MALIDUP-ns集的散点图。 左下方显示MALISAM-ns设置。
图6显示了从MALISAM-ns测试集中获得的最困难比对之一的一个例子。该对中的一个是甲酰转移酶 - 环丁胺酶的甲氨基转移酶结构域的结构(如左图6a所示)。 另一种是FAD结合结构域的置换结构(图6a右),其通过随机排列FAD结合结构域的SSE的连续顺序而产生。
参考比对的比对图和九种比对方法的比对图也显示在图6b和c中。 参考的结构对齐由五个片段组成,每个片段包含一个SSE和连接到SSE的环。 多肽链中SSE的顺序完全不同,这使得比对非常复杂,如图6b所示。 五个非顺序方法(SNAP,GANGSTA +,SCALI,MASS和SAMO)与参考比对返回零一致,表明难以再现该对的参考比对。 DaliLite,TM-align和DEDAL与参考文献返回部分一致。 所有这三种方法都正确地将α 2与α 2对齐,但只有DaliLite将α 1与α 1进行了对比。除MICAN之外的所有方法都未能正确对齐任何β链对。 另一方面,只有MICAN正确对齐所有二级结构对,完全再现参考对齐(Q-score = 100%)。
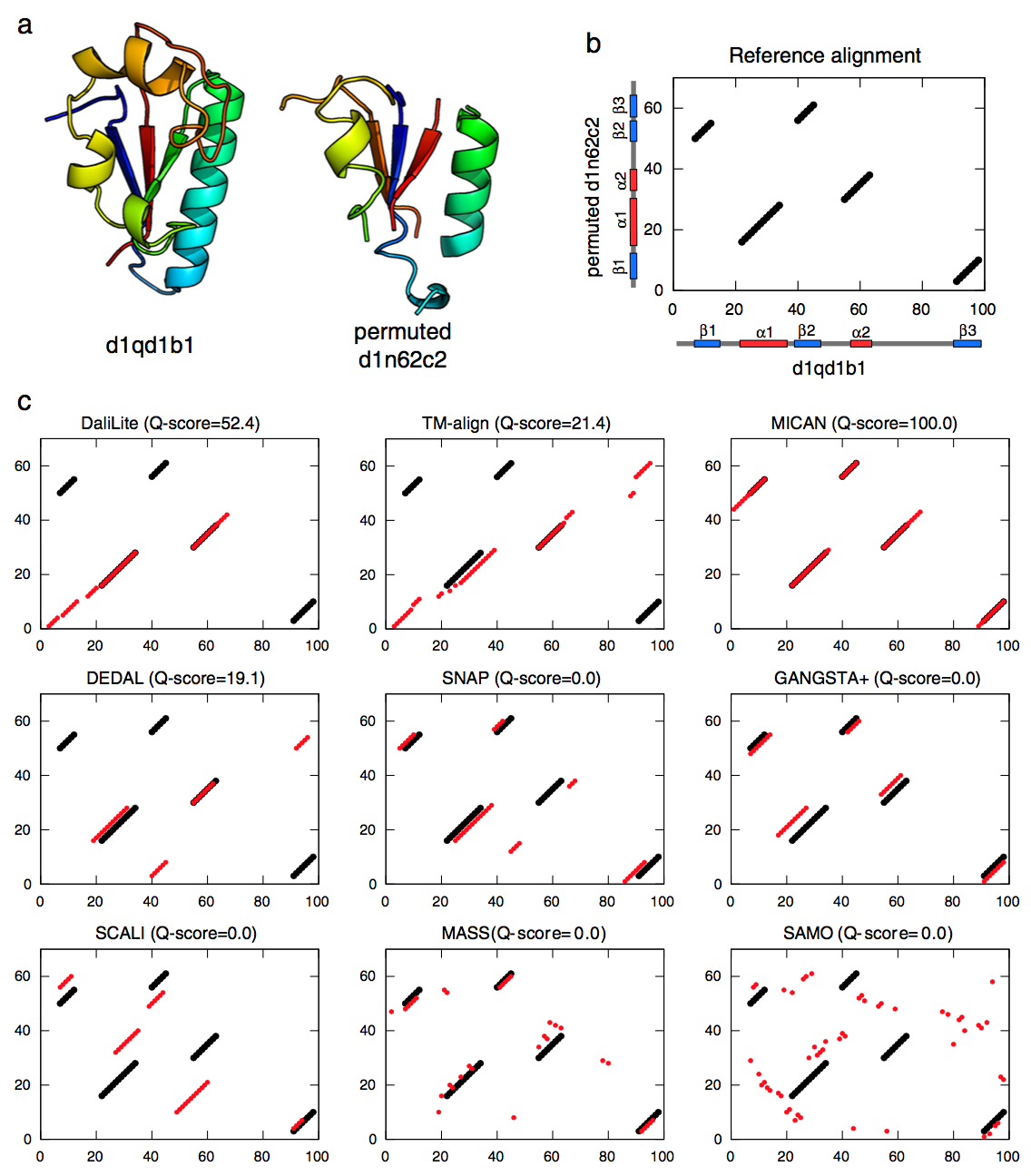
图6 从MALISAM-ns集中获取的难以非顺序对齐的示例。 (a)甲酰基转移酶 - 环丁胺酶(SCOP-ID d1qd1b1,B2083-B2180)的甲氨基转移酶结构域和FAD结合结构域的置换结构(SCOP-ID d1n62c2,C1-C61)。 (b)参考比对的对齐图。 横轴和纵轴分别对应于d1qd1b1的残基位置和置换d1n62c2的残基位置。 α-螺旋和β-链在天然结构中的位置也用红色和蓝色条表示。 (c)比较参考比对和我们在此测试的九种比对方法之间的一致性。 在每个比对图上显示了方法的名称及其蛋白质对的Q-分数。 参考对齐对以黑色圆圈显示。 每种方法生成的对齐对以红色圆圈显示。
尽管MICAN在参考依赖性评估方面是最佳的比对工具,但仍然存在一些失败。 例如,如图3和5所示,MICAN生成一些Q-score = 0%的比对。 人工调查显示,大多数此类失败的路线都具有共同特征; 它们相对于参考比对具有1-4个残基位移。 图7所示是MICAN的典型失败示例。 虽然MICAN的对齐图类似于参考图,但是没有与参考对齐重叠。 MICAN(0.503)对齐的TM得分略大于参考(0.498)的对比,表明优化TM得分并不总是与人工比对的一致。 这一观察结果表明,一种可能的改进是开发一种与人类专业知识更加一致的评分函数。(有的时候,人工调整,比对出来的更准确呗? )

图7 MICAN失败对齐的示例。(a)与参考比对(黑色)和MICAN(红色)比较。 该蛋白质对(d1ovma1和置换d1914a2)取自MALISAM-ns。 横轴和纵轴分别对应于d1ovma1的残基位置和置换d1914a2的残基位置。 α-天然结构中α-螺旋和β-链的位置也用青色和粉红色条表示。 (b)根据参考对齐的叠加。 d1ovma1的结构用蓝色和青色着色,而置换的d1914a2的结构用红色和粉红色着色。 蓝色和红色区域表示对齐的残基,青色和粉红色表示未对齐的残基。 (c)基于MICAN对齐的叠加。
3.5 基准reference-independent评估的基准测试
为了识别几个比对程序的特征,对于所有方法生成的所有对齐,对于MALIDUP-sq,MALISAM-sq,MALIDUP-ns和MALISAM-ns测试集中提供的所有对,我们计算了几个标准几何度,对齐残基对的数量(Nali),Cα原子的相应均方根偏差(RMSD)和间隙开口数(Ngap) 。 这里,Nali定义为比对的残基对的数量除以蛋白质对的较小蛋白质的大小。 它们的MALIDUP-sq,MALISAM-sq,MALIDUP-ns和MALISAM-ns测试集的平均值列于表2中。为了可视化数据,通过每种方法获得的平均Nali和RMSD绘制在(Nali,RMSD)图8。在此平面上,更好的性能对应于,沿水平轴的较大值和沿垂直轴的较小值。

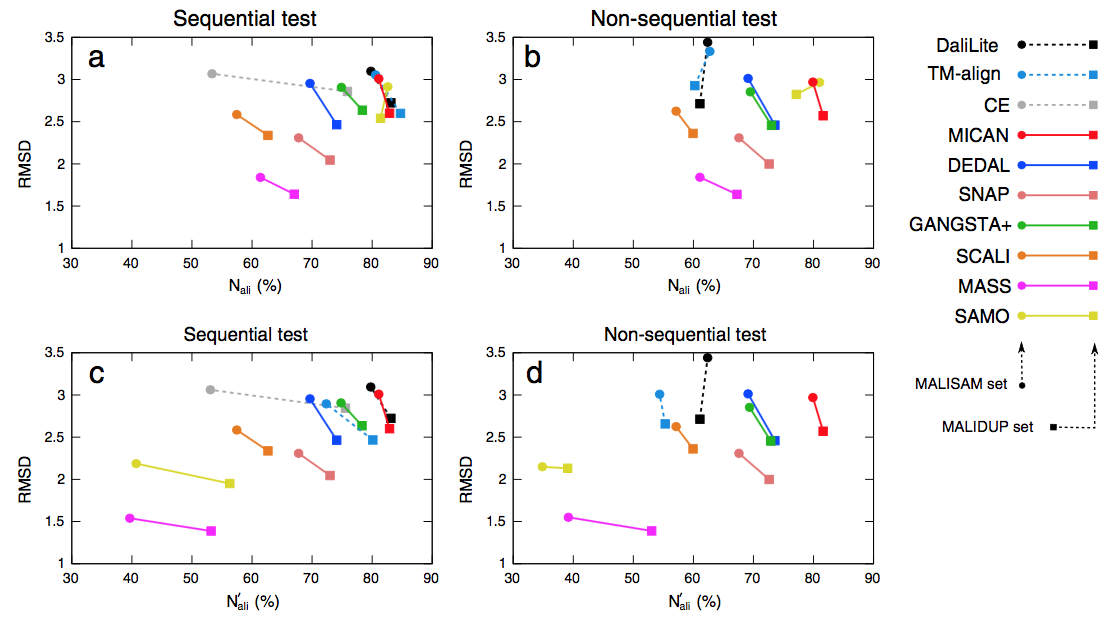
图8 reference-independent 评估的结果。 对于所有测试(引导视线的线条),在(Nali,RMSD)平面上绘制参考独立的结构比对评估结果。 X轴和y轴分别表示标准化的排列残基数及其RMSD。线颜色代表程序:黑色(DaliLite),青色(TM-align),灰色(CE),红色(MICAN),蓝色(DEDAL) ),三文鱼(SNAP),绿色(GANGSTA +),橙色(SCALI),紫色(MASS)和黄色(SAMO),线条样式代表程序的类型:虚线表示顺序对齐程序,而实线表示非顺序对齐程序。 基准集由符号表示:square(MALIDUP-sq / ns)和circle(MALISAM-sq / ns)。 (a)和(b)分别显示Nali和RMSD之间的顺序和非顺序测试集之间的关系。 (c)和(d)分别显示了连续和非连续测试集的Na’li和RMSD之间的关系。
图8a所示为MALIDUP-sq和MALISAM-sq测试集的结果。 每种方法都位于平面的不同位置,这反映了每种方法的目标函数的特征。 其中,DaliLite,TM-align,MICAN和SAMO显示出相似的排列特征; 他们位于类似的位置。 这四种算法位于图的右上部分,表明它们更喜欢具有较大RMSD(约3Å)的较大数量的对齐残基对。 虽然它们的平均RMSD值与其他程序相比相对较大,但这些值可以被认为是生物学相关的,因为它们与同源蛋白质的典型RMSD相当[39]。 相比之下,MASS的对齐显示与所有算法的最小RMSD的较短对齐。 其他程序介于两种极端情况之间。
图8b显示了MALIDUP-ns和MALISAM-ns测试集的结果。 与图8a相比,正如我们所预期的,Nali的顺序算法显着减少。 另一方面,与顺序算法相比,平面上所有非顺序算法的位置不受从顺序测试集到非顺序测试集的测试集的变化的影响。 特别是,显示MICAN,DEDAL,SNAP和MASS对于序列顺序的改组非常强大。 他们在Nali和RMSD的变化不到2%。
参考资料
- 《MICAN : a protein structure alignment algorithm that can handle Multiple-chains, Inverse alignments, C α only models, Alternative alignments, and Non-sequential alignments》 https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-14-24
Å
