【1.1.1】蛋白溶剂可及性(solvent accessibility)
可及表面积(ASA,accessible surface area)或溶剂可及表面积(SASA,solvent-accessible surface area)是溶剂可接触的生物分子表面积。 ASA的测量通常以平方埃为单位进行描述(分子生物学中的标准测量单位)。 ASA由Lee&Richards于1971年首次描述,有时被称为Lee-Richards分子表面。[1] ASA通常使用Shrake&Rupley在1973年开发的“滚球”算法来计算。[2] 该算法使用特定半径的(溶剂的)球体来“探测”分子的表面。
与范德华表面相比,溶剂可及表面的图示。 由原子半径给出的范德华表面以红色显示。 可触及的表面用虚线绘制,并且是通过沿球体范德华表面滚动跟踪球体的中心(蓝色)而创建的。 请注意,此处描绘的探针半径的比例尺小于典型的1.4Å。

二、计算溶剂可及性
溶剂分子在蛋白质的范德华表面上滚动时的中心,如图3.2a所示(Gromiha和Ahmad,2005)。 通常,假定水球是半径为1.4 Å的溶剂分子。 溶质分子由分配给每个原子的适当范德华半径的一组互锁球表示(interlocking spheres),溶剂分子沿范德华表面的外壳在方便剖切的平面上滚动。 因此,半径为r的原子的ASA是半径为R = r + rsolv的球体表面上的区域,在该区域的每个点上,可以使溶剂分子的中心与该原子接触,而不会穿透任何其他原子 溶质分子。 溶剂ASA使用以下公式计算(Lee和Richards,1971):

图3.2。 (a)溶剂可及性的定义。 显示了探针半径,范德华表面和可及表面。 该图改编自Gromiha和Ahmad(2005); (b)由两个平面切出的孤立范德华球面的二维表示。 标记了溶剂可及区域计算中涉及的参数。
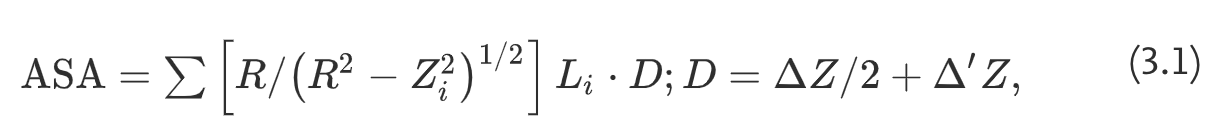
其中Li是在给定截面i上计算的弧的长度,Zi是从球体中心到截面i的垂直距离,Z是截面之间的间距,并且’ZisΔZ/ 2或R-Zi, 以较小者为准。 给定原子绘制的所有弧线上的求和。 图3.2b显示了由平面i和i + 1切割的孤立原子的相关参数。
已经开发了一些程序,用于从蛋白质和复合物的三维结构中计算原子和残基的ASA。 本节介绍了所选方法的详细信息。
2.1 ACCESS
Richmond和Richards(1978)引入了用于计算ASA的程序ACCESS。 该程序采用蛋白质/核酸的三维结构,并计算每个原子的可及和接触表面积。 它使用PDB的默认设置接受不同类型的输入格式。 ACCESS使用的标准探针半径为1.4 Å ,并且根据原子名称和残基名称分配原子的范德华半径(Lee and Richards,1971)。 ACCESS具有以下不同选择:(i)计算原子的面积,(ii)在可访问性计算过程中忽略原子,以及(iii)原子被认为是蛋白质环境的一部分,因此不会对该原子进行任何计算。 ACCESS的最终输出给出每个原子的ASA,每个残基的主链原子的总ASA,每个残基的侧链原子以及蛋白质的总ASA。 可从 http://www.csb.yale.edu/ 获得访问权限。
2.2 Naccess
Hubbard和Thornton(1993)开发了一个程序Naccess,用于计算原子可及区域。 该程序使用Lee和Richards(1971)的方法,其中将给定半径的探针绕分子表面滚动,并且以其中心绘制的路径为可访问表面。 Naccess是一个独立程序,可从PDB格式文件计算分子的可访问区域。 它可以计算蛋白质和核酸的原子和残基可及性,可供研究人员免费使用。 该方法的主要特征是可以在用户定义的探针大小,残基(氨基酸,核酸和杂原子),原子半径,标准状态残基可及性以及有/无杂原子,氢和水的情况下运行。 它将生成原子ASA的输出,绝对和相对残差ASA以及计算日志文件。 Naccess可从 http://wolf.bms.umist.ac.uk/naccess/ 获得。
2.3 GETAREA
Fraczkiewicz和Braun(1998)开发了GETAREA程序,用于在网络上计算生物大分子的ASA。这是一个高效的程序,它使用户可以提交分子中原子的笛卡尔坐标,以PDB格式存储,并以多种格式获取ASA或溶剂化能量(取决于参数设置)。默认情况下,提交表单设置为计算蛋白质中非氢原子的ASA,但是输入参数的适当更改将允许针对任何种类的分子计算与ASA成比例的任何数量。它具有用于计算和显示每个原子,原子类型,残基和总面积的ASA的选项。图3.3说明了不同类别的输入选项(图3.3a)和输出结果。该程序的输出为每个原子(图3.3b),原子类型(图3.3c),残基(图3.3d)和整个蛋白质(图3.3e)提供了ASA。 GETAREA可从 http://www.scsb.utmb.edu/getarea/ 获得。
2.4 DSSP
Kabsch和Sander(1983)开发了一种DSSP方法,以计算残留物的溶剂可及性(图3.1a),并为ASA存储了PDB中大多数蛋白质的数据库(Berman et al.2000)。 生成用于预测算法的ASA值的常用程序/数据库之一(Rost和Sander,1994; Pascarella等人1998; Mucchielli-Giorgi等人1999; Ahmad等人2003; Gianese等人2003; Qin 等人,2005; Momen-Roknabadi等,2008)。 DSSP可从 http://www.cmbi.kun.nl/gv/dssp/ 获得。
2.5 ASC
Eisenhaber和Argos(1993)开发了一个程序包,分析表面计算(ASC),用于计算蛋白质/核酸中每个原子和残基的ASA。 ASC在程序中具有以下选项:(i)通过新的分析方法计算一组相交球的表面(范德华表面或具有任何探针半径的可接触溶剂的表面),(ii)数值确定 通过双立方晶格法测定表面的质量,以及计算体积和点表面,以及(iii)计算分子及其组成部分的表面能和亲水/疏水表面。 该程序用C语言编写,可供研究人员下载。 ASC为每个原子和残基提供不同格式的输出和ASA。 可从以下网址获得ASC: http://mendel.imp.univie.ac.at/mendeljsp/studies/asc.jsp 。
2.5 POPS
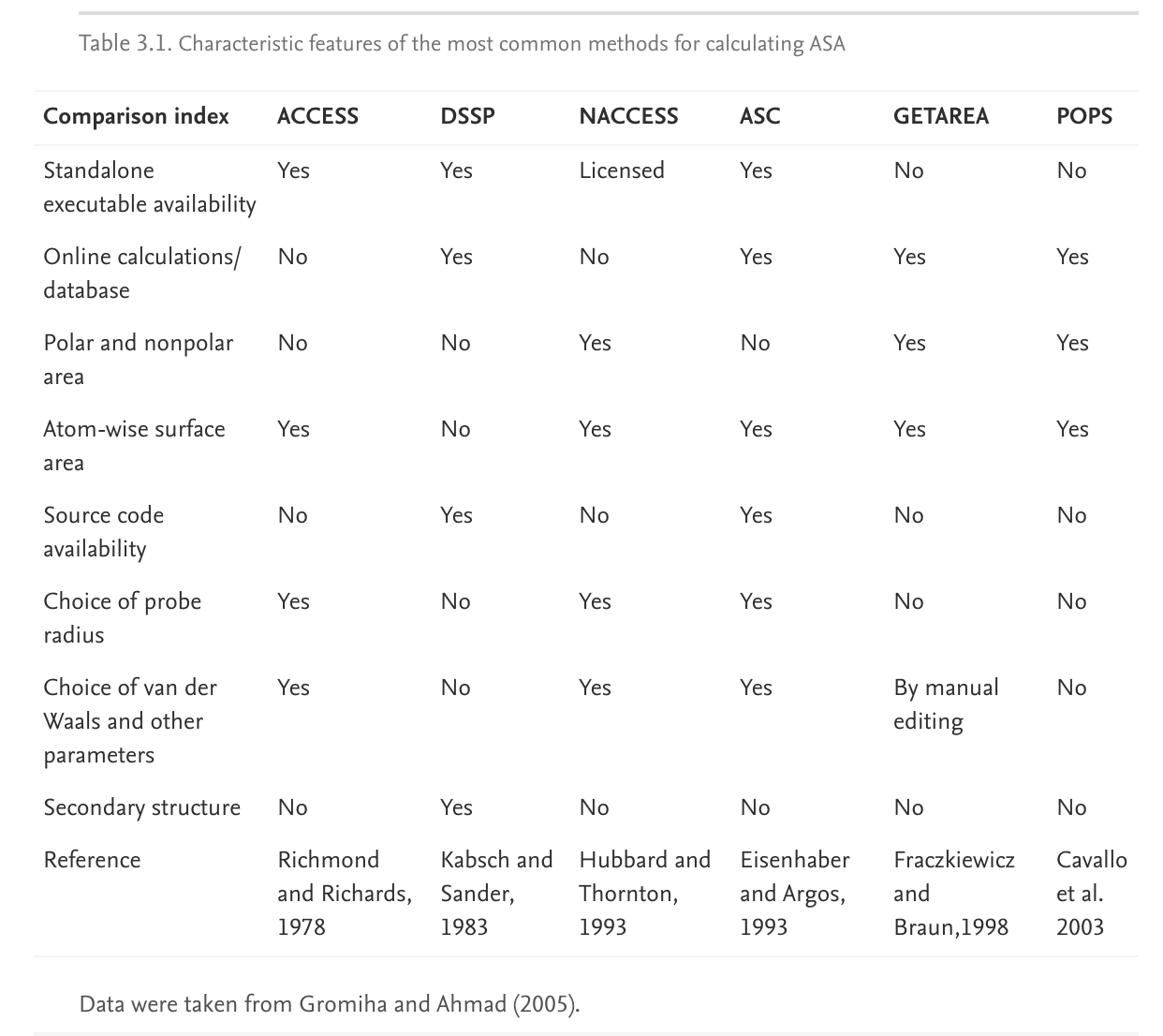
Cavallo等(2003年)开发了一种方法,即POPS(参数优化表面),该方法基于经验可参数化的分析公式来计算原子级和残留级溶剂ASA。 参数化来自具有不同大小和拓扑结构的选定蛋白质数据集。 可以在 http://mathbio.nimr.mrc.ac.uk/wiki/POPS和镜像站点www.cs.vu.nl/∼ibivu/programs/popswww 上获得POPS程序。 表3.1比较了上面列出的最常用程序之间的主要相似点/不同点。
参考资料
