【6.5.1】从头对接,建模和合理设计的实验指导计算抗体亲和力成熟
抗体是一类重要的疗法,对严重疾病的治疗具有重要的临床影响。在过去的十年中,支持抗体药物发现的计算工具正在以越来越高的速度发展,并且通常依靠与抗原结合的抗体的预定共晶体结构来进行结构预测。在这里,我们展示了一个杂交瘤衍生抗体AB1成功进行计算机亲和力成熟的例子,仅使用抗体片段可变区的同源模型和与抗原鼠CCL20结合的AB1抗体的蛋白质-蛋白质对接模型( muCCL20)。在计算机亲和力成熟中,与丙氨酸扫描一起,使我们可以微调蛋白质-蛋白质对接模型,从而随后鉴定出两个单点突变,从而增加了AB1对muCCL20的亲和力。据我们所知,这是使用同源性建模和蛋白质对接进行亲和力成熟的第一个例子,并且代表了可以广泛使用的方法。
作者总结
计算技术在治疗性蛋白质开发中的作用是多方面的,包括结构预测(同源性建模),界面识别(对接)和突变能变化计算。据报道,在蛋白质结构预测和界面预测领域取得了成功(参见竞争结果,例如结构预测的关键评估[CASP]和预测相互作用的关键评估[CAPRI]),但是最大的挑战之一可能是突变后,计算机合成的结合能转化为亲和力成熟的抗体变体。在这些应用中,重要的是选择在计算机蛋白质设计的各个方面都有意义的正确结构模型或近似模型。当没有抗体-抗原共晶体结构可用,并且蛋白质-蛋白质界面周围存在高度不确定性时,挑战就变得更加复杂。尽管可以说该领域距离其将计算预测与实验数据精确关联的目标相去甚远,但我们表明,即使在不存在共晶结构的情况下,也可以通过计算机诱变技术与同源性建模,蛋白质对接和简单的实验检查点。
一、前言
抗体是已知的最特殊的结合分子类型,其多功能性已导致许多成功的治疗严重疾病的疗法。 在结构上,抗体是由β-折叠形成的多域蛋白,这些折叠通过二硫键连接在一起。 将两个免疫球蛋白结构域(可变轻链(VL)和可变重链(VH)域)连接在一起以创建可变片段(Fv)。 Wu和Kabat的原始工作[1]在VH和VL结构域中确定了六个高变区,并正确地预测了这些区域是抗原特异性结合的原因。 这些环,即互补决定区(CDR),来自相对保守的框架区(FR),通常在空间上与抗原非常接近。 VL和VH结构域共同产生抗原的结合位点,该结合位点大部分由CDR介导。
抗体发现平台使用基于展示的文库方法(噬菌体,酵母,核糖体,哺乳动物或其他系统)或用于抗体分离的免疫和杂交瘤筛选策略。 一旦分离出一组先导抗体,如果抗体要成为潜在的治疗药物,它们的结合亲和力通常需要优化。 上述展示方法可用于体外亲和力成熟,因为它们可以控制抗原浓度,呈递形式和取消选择以消除不需要的特异性。 这些方法以及其他随机诱变方法已被证明非常有效地提高了亲和力[2-7]。 然而,体外亲和力成熟的过程可能是费力且耗时的,需要花费数月的时间,并且提高亲和力的更有效方法将是有益的。
已经报道了许多用于计算机抗体亲和力成熟的策略,通常采用基于
- 结构的原理[8-11]
- 或小型文库方法[12]。
这些方法的成功主要取决于两个因素:
- 第一,高质量共晶结构的存在,
- 第二,计算突变时发生的能量变化的算法。
在文献中已经报道了应用自由能扰动(FEP,free energy perturbation )[13-15]和平均势力(PMF,potential-of-mean force )[16]方法来预测蛋白质的自由能变化[17-20]。但是,它们通常需要大量的计算时间和成本,这显着限制了它们在计算机抗体lead优化中的应用。更常见的是,基于分子力学(MM,molecular mechanics )的方法,再加上隐式溶剂模型,例如广义Born表面积(MM-GBSA,generalized Born surface area),分子力学-Poisson-Boltzmann表面积(MM-PBSA,molecular mechanics-Poisson-Boltzmann surface area)[21,22]或使用Lazaridis-Karplus溶剂化模型(MM-LKSM)来估计自由能,从而显着节省了计算时间和基础设施[23-26],从而可以计算数千个突变,而不是数十个突变。使用分子力学计算的缺点是它们不能解释突变后可能出现的整体构象变化,从而导致ΔΔE值不能真正代表真实蛋白质。 MM-GBSA / MM-PBSA / MM-LKSM自由能变化与实验数据之间的相关性很差,这通常小于0.5(皮尔森相关性)[27,28]。
拥有高质量抗体结构信息被广泛认为是计算抗体亲和力成熟的必要起点。据我们所知,关于计算机亲和力成熟的公开报道很少,其中抗体-抗原复合物的共晶体结构不可用。在这种情况下,由于在大型生物系统的计算和模拟过程中会传播许多假设和错误,因此亲和力成熟尤其具有挑战性,特别是如果野生型抗体已经是纳摩尔摩尔范围内KD的强结合剂时。尽管存在这些挑战,我们还是决定测试这种方法,选择建立Fv片段的同源性模型,并使用抗原的核磁共振(NMR)结构进行蛋白质-蛋白质对接。我们从实验组的VH和VL域中的丙氨酸扫描突变的实验指导中优化了蛋白质-蛋白质对接,并使用了计算机模拟复合物来驱动计算机模拟亲和力成熟。在本文中,我们是第一个描述这种方法为获得高亲和力抗体的亲和力和活性而获得的方法的应用,该方法虽然相对适度,但却证明了这种方法的潜力。
二、结果
2.1 小鼠CCL20-AB1的同源性建模和蛋白对接
结构准备。
选择鼠蛋白CCL20(muCCL20)及其结合物AB1作为我们的测试用例,因为它们符合纳摩尔范围结合亲和力以及试剂和分析可用性的标准。 由于唯一可用的晶体结构是抗原的晶体结构,而不是抗原-抗体复合物的晶体结构,因此我们有机会通过同源性建模研究从头进行亲和力改善的潜力。 AB1 Fv模型由SabPred [29]生成(图1A),而muCCL20的结构来自蛋白质数据库(PDB ID,1HA6)并用于对接(图1B)。
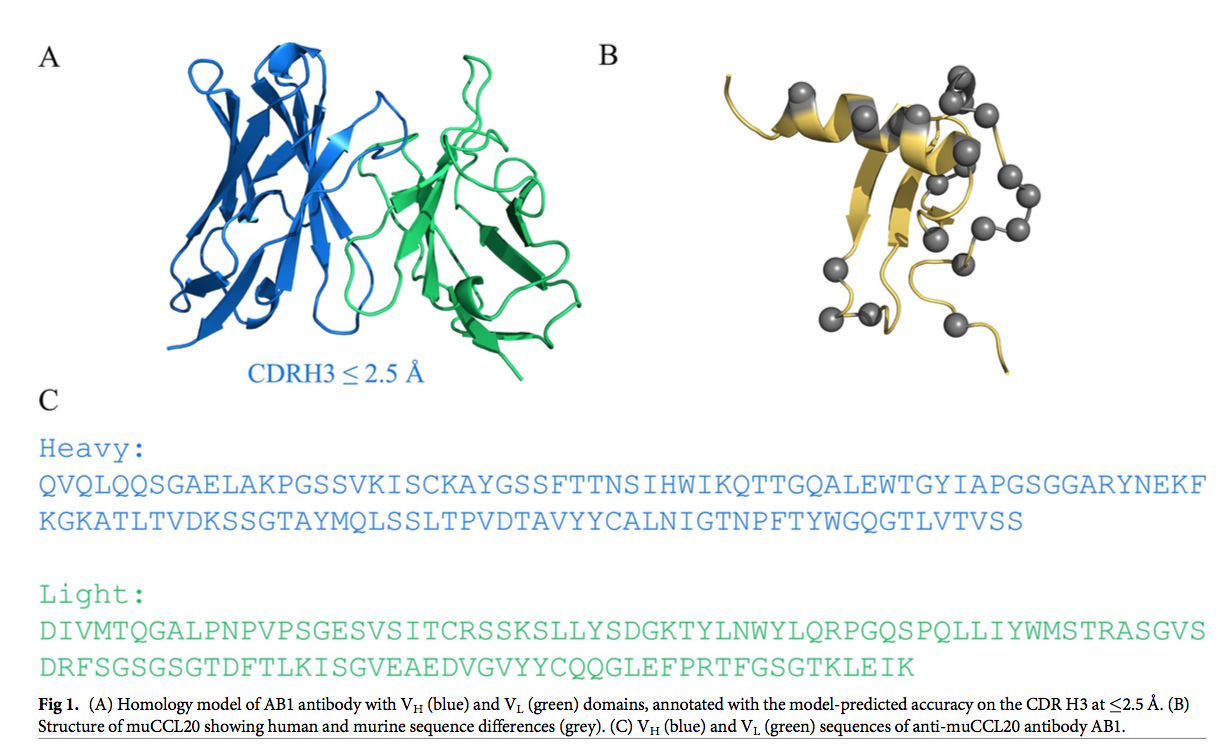
蛋白质-蛋白质对接用于建立CCL20和AB1 Fv区之间的假定复合物。 对接过程总共产生了54,000个姿势,其中前2,000个poses被进行了改进。 这2000个姿势用RDOCK算法进行了改进[29]。 RDOCK得分排名前5位的姿势都包含在三个结构簇中。 前五个姿势中的每个姿势的RDOCK得分均小于–30.00,最佳姿势中的得分为–36.20(表1)。 溶剂可及表面积(SASA, Solvent accessible surface area)用作结合后掩埋的表面积的量度。
在我们的案例研究中,抗原muCCL20与人类直系同源物具有67%的序列同源性(图2A)。 相比之下,muCCL20与病毒巨噬细胞炎性蛋白2(vMIP-II)之间的序列同一性为30.65%,序列相似性为48.39%。 我们进行了结合酶联免疫吸附测定(ELISA),发现我们的抗体AB1不结合人CCL20(图2B),这为我们提供了关键数据,以帮助确定可行的对接姿势。 此外,我们进行了基于细胞的活性分析,其中通过muCCL20与细胞表面表达的受体CCR6相互作用而赋予的活性被AB1阻断。 因此,有可能进一步将对接搜索空间缩小为仅包含那些阻止此交互的姿势。
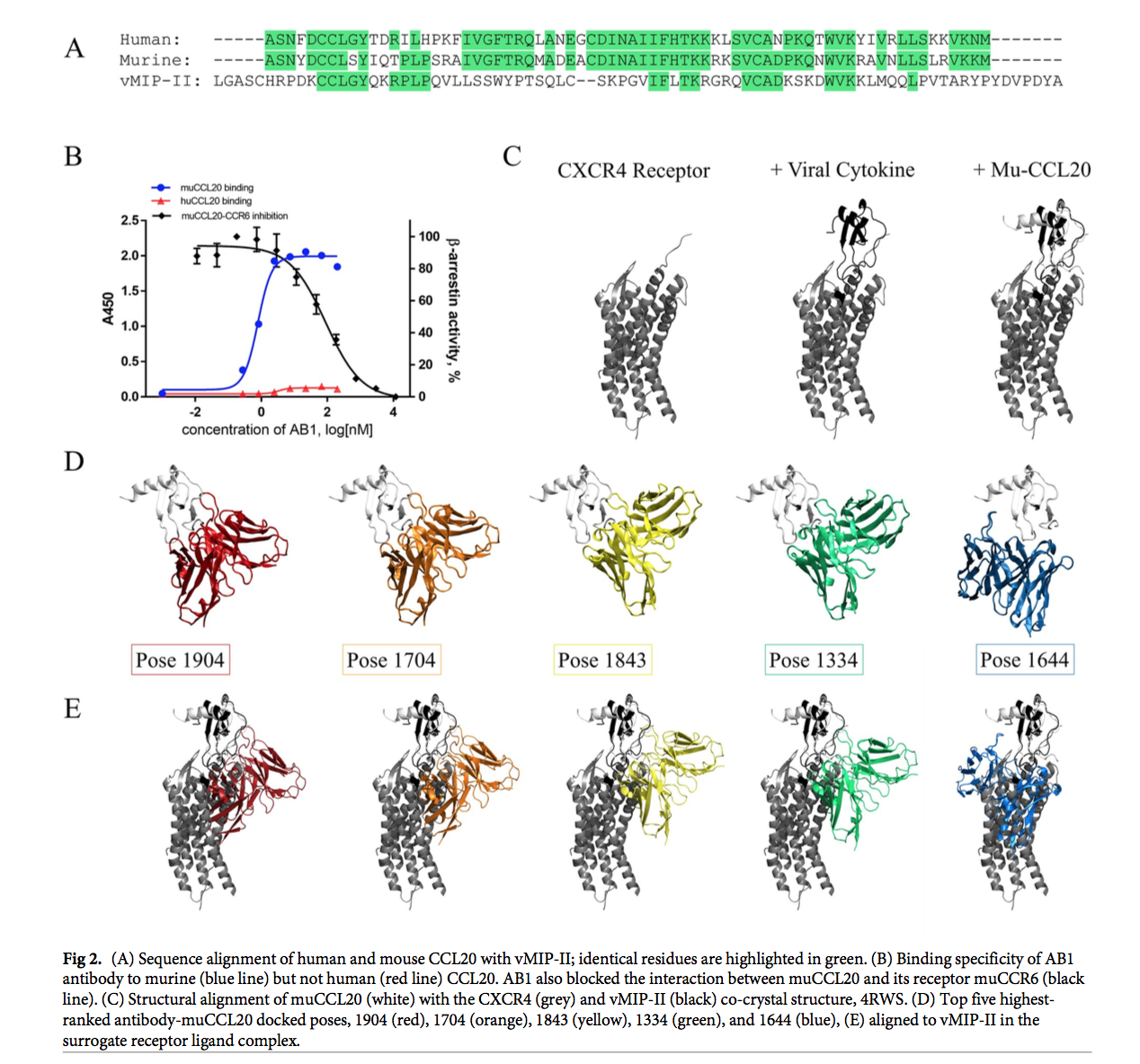
在撰写本文时,尚无人或鼠CCR6的晶体结构,无论是作为受体还是单独作为受体,都难以评估配体-受体相互作用的立体特异性阻断。 因此,为了指导对接实验,我们从代理复合体中推断出结构知识。 受体和趋化因子折叠在CXCR受体家族中都非常保守,因此我们使用另一个受体-配体家族成员人CXCR4和vMIP-II(PDB ID,4RWS)的共晶体复合物作为模板 CCR6-CCL20复合物的构象(图2C)。 使用jFATCAT [30](刚性)比对算法,发现两个结构的均方根偏差为2.35Å。
将每个顶部对接的姿势与CXCR4-vMIP-II复合物对齐,以确定相对的抗体-抗原构象(图2D)是否满足立体特异性阻断细胞因子-受体结合的标准(图2E)。 在五个对接姿势中的每个姿势中,AB1 Fv都被受体覆盖,这表明这些姿势将潜在地阻止CCL20与CCR6受体的相互作用。
为了了解每个姿势的结合特性,我们进行了界面分析并确定了抗体与抗原之间明确的分子间相互作用的数量(图3)。 排名最高的姿势(1904)和排名第三的姿势(1843)都显示出最多的分子间接触。 从muCCL20向人CCL20的突变同时进行,导致每个姿势的结合亲和力丧失(S1表); 最大的相对损失归因于1843位,它具有最大的总分子间相互作用数量(+2.57任意单位[AU])。 抗muCCL20抗体不会与人抗原发生交叉反应,因此,为了进一步评估前五个姿势的有效性,我们对muCCL20进行了计算机诱变,预计将产生最终的“物种转换”(species switch) 失去抗体的亲和力。
Refining the structural model of AB1 by alanine scanning in silico. 在计算机上通过丙氨酸扫描完善AB1的结构模型。
我们对前五个姿势进行了计算机丙氨酸丙氨酸扫描,以通过鉴定对不同蛋白质-蛋白质结合构象的亲和力很重要的残基进一步完善模型。 五个姿势中的每个姿势均具有独特的丙氨酸扫描曲线(图4),该功能可用于通过实验验证对接过程并识别最接近真实复杂构象的对接姿势。 选择用于实验验证的突变体是基于三个标准:
- 必须包含在CDR(国际ImMunoGeneTics信息系统[IMGT] [31])loop中,包括游标(Vernier)残基;
- 参与抗体和抗原之间独特的,明确的分子间相互作用;
- 与其他候选姿势中的至少两个相比,突变为丙氨酸时ΔΔE结合值存在显着差异。
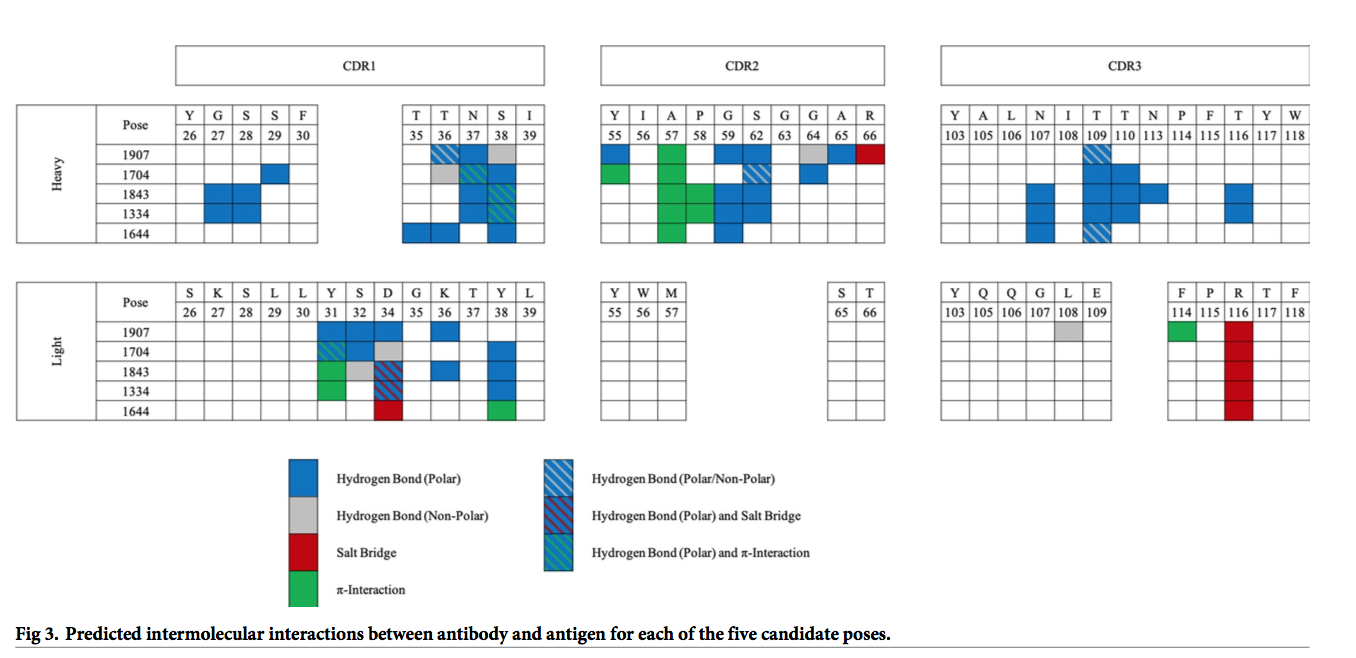

使用这些标准,我们确定了四个单点突变,针对这些突变预测了抗体和抗原之间的明确分子间相互作用,并且这是五个姿势之一所独有的。 选择了另外9个单点突变,并选择了5个三重突变作为单个突变的组合,每个突变在ΔΔE结合值上表现出显着差异。 最后,选择了两个阴性对照突变,每个抗体链上一个,其中丙氨酸突变后ΔΔE结合值没有变化,共有20个变体(表2)。

2.2 小鼠CCL20-AB1的蛋白对接的实验指导优化 Experimentally guided refinement of protein-protein docking of murine CCL20-AB1
关键抗原-抗体接触残基的实验鉴定
进行了定点诱变,以产生涉及抗体-抗原相互作用的可能关键残基的丙氨酸突变。总共产生了23个变体,包括表2中详述的20个变体,以及在VH和VL突变组合期间作为中间结构生成的3个其他变体(变体21,L:Y31A / Y38A;变体22,L:Y31A / D34A;以及变体23,H:T35A / N107A / L:Y31A)(图5)。为了区分这些突变体之间的结合差异,我们使用捕获ELISA评估了抗原结合,以更好地近似于1:1结合,也就是说,使用板包被的抗碎片可结晶(Fc)抗体捕获了一组针对muCCL20结合的抗体变体。在这23种变体中,有11种完全失去与muCCL20抗原的结合,而3种(变体5、7和16)则显示结合力降低,而其余变体则保持结合。鉴定为单点突变的非结合变体有助于我们剖析参与结合相互作用的关键残基,而双突变和三突变则进一步证实了这一点。对结果进行反卷积(Deconvolution)显示,H:Y55,H:N107,H:N113,L:D34和L:Y38这五个位置对于抗原结合至关重要,因为丙氨酸置换会导致信号完全消失,并且该位置L:K36也参与其中,因为丙氨酸突变使结合亲和力降低了三倍(图5)。

结合ELISA数据与计算机诱变结果的比较发现,这五个候选位点均未与实验数据完全一致。对于显示减少或没有结合的14个变体中的每一个,最多损失18个分子间相互作用。在每个姿势上进行结构分析,以确定抗体和抗原之间是否失去了特定的分子间相互作用。姿势1843显示出最大的相关性。丙氨酸突变会丢失18种分子间的相互作用中的16种,并且姿势1334(13个相互作用),1704(11个相互作用),1904(9个相互作用)和1644(8个相互作用)之间的相互作用与实验数据的相关性较小。这些发现的含义是,尽管位点1843确实确实确实最接近于天然结构,但它不满足所有“非结合”变体标准,因此在计算机亲和力成熟中不可靠。因此,为了提高结构模型的可靠性,我们使用这些实验诱变数据来指导设计,从而对抗体和抗原进行了对接。
####Redocking of antibody and antigen.
重复蛋白质对接,并将残基H:Y55,H:N107,H:N113,L:D34,L:K36和L:Y38定义为在抗原5.0Å以内的界面。随后改进了1,925个姿势。不仅仅是简单地按得分选择排名最高的姿势,而是对每个姿势进行单个丙氨酸突变,以识别与导致结合减少或丧失的突变的实验结合谱相匹配的突变。一组10个单丙氨酸突变-5个非结合和1个减少的结合变体(H:Y55A,H:N107A,H:N113A,L:D34A,L:K36A和L:Y38A)和4个对照(H :R66A,H:T110A,L:Y31A和L:R116A)—被设计成1925所有对接姿势。从计算机诱变实验中,对ΔΔEbinding值进行编译和过滤,以便仅当六个有害突变的ΔΔEbinding值≥0.00AU并且四个中性突变的ΔΔEbinding值≤0.50AU时才接受姿势。 1925只有8个构象符合所有标准(S2表)。
因为L:K36A突变导致结合减少而不是完全丧失,所以相对ΔΔE结合值应小于其他有害突变。 姿势491(在整体上排名第120)比我们确定的其他姿势更匹配此配置文件。 随后选择该模型进行计算机亲和力成熟(图6)。

2.3 Affinity maturation of AB1 through in silico design and experimental validation 通过计算机设计和实验验证AB1的亲和力成熟
In silico affinity maturation.
为了最大化识别可改善抗体-抗原结合亲和力的突变的机会,我们使用了三种独立的计算机模拟算法来进行诱变:Discovery Studio 2016 [32],SchrödingerBiologics Suite 2016-3 [33]和Rosetta [34] ]。 可设计的残基仅限于Kabat[1]和IMGT [31] CDR残基的杂种选择,以最大化可变区中的序列空间,同时避免构架残基。 CDR定义为CDRH1:G27-H40,CDRH2:Y55-G74,CDRH3:A105-Y117,CDRL1:R24-N40,CDRL2:W56-S69和CDRL3:Q105-T117(IMGT编号)。
为了确保仅识别出最主要的突变,在≤1.0 AU的ΔΔE结合值和≤0.5 AU的ΔΔ稳定性值的基础上,对Discovery Studio和SchrödingerBiologics Suite中的突变进行了过滤。在1,400个Discovery Studio突变中,有46个满足了准则,其ΔΔEbinding值在-2.46至-1.02 AU之间。将筛选器应用于两组Schro-dinger突变(0.0-和5.0Å优化半径),并接受针对0.0Å协议的87个突变和针对5.0Å协议的154个突变。两种方案的共识显示了这两种突变共有的28种突变,对于0.0Å最适半径截止值,ΔΔEbinding值范围从-18.75至-1.12,对于5.0Å截止值具有-12.13至-1.20。根据Rosetta的逐个站点优化协议,发现29个站点具有可接受的突变。通过从突变体ΔE结合值中减去野生型ΔE结合值并确定最稳定的突变来计算结合能值。此外,将每个接受的突变与点突变稳定化结果进行交叉检查,以确保该突变不会使复合物不稳定,最终鉴定出19个突变。
在这三种方法中鉴定出的93个潜在突变中,鉴定出6个重复,将panel减少到87个。选择了20个突变的panel(表3)在实验室中进行经验测试。 这20个在VH和VL结构域中包含了不同范围的界面位置,并且在所采用的三种方法中都包含了最有潜力的有益突变。 我们还检查了没有排除预测的热力学稳定的水分子。 最后一组包括用Discovery Studio鉴定的4个突变,用Schro dinger鉴定的11个突变,用Rosetta鉴定的4个突变和根据所有这三种方法预计不会产生影响的1个对照。

结合和活性测量,以鉴定亲和力成熟的变体。
通过使用定点诱变构建了20个预测的变体,VH结构域中的13个点突变和VL结构域中的7个点突变的组,随后进行表达和纯化。捕获酶联免疫吸附法(ELISA)显示,两个变体,克隆1(或AB1-C1,H:S28R)和克隆16(或AB1-C16,L:G35Y),在结合EC50上显示出明显的变化,而其余的突变体与亲本相比,保留或减少了结合。我们随后将VH和VL突变组合到一个组合克隆中,克隆1–16(或AB1-C1-16),发现它在结合曲线中保持了左移(图7A)。为了进一步证实这些变体确实赋予了改善的结合亲和力,我们着手用木瓜蛋白酶进行蛋白水解,从这些纯化的抗体中生成Fab片段。通过与Biacore的表面等离振子共振来测量1:1的结合动力学和亲和力。与亲本AB1相比,克隆AB1-C1的结合解离常数(KD)改善了4倍,而AB1-C16的结合KD改善较小。组合克隆AB1-C1-16的行为类似于AB1-C1。
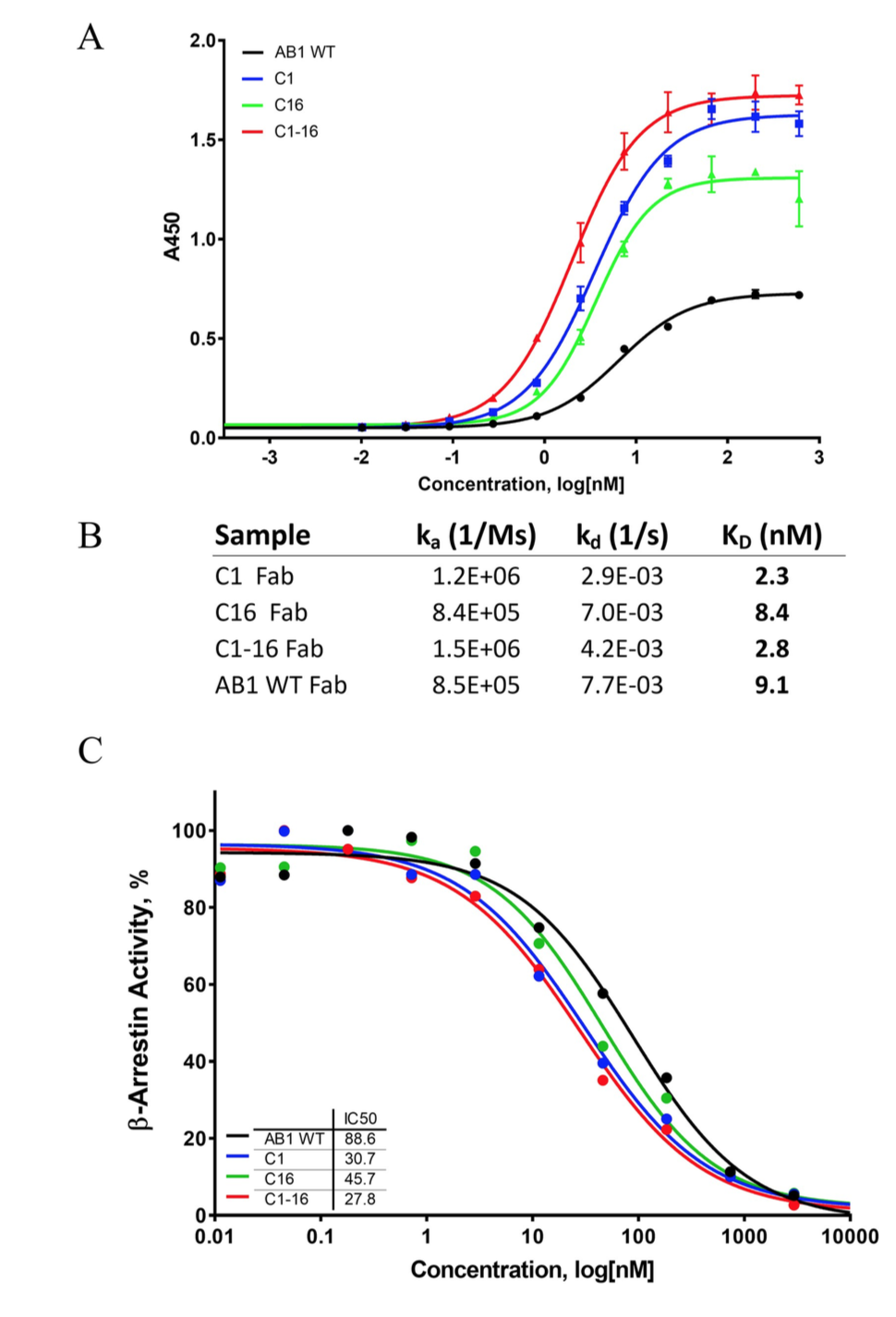
Fig 7. (A) ELISA revealed improved antigen binding with VH clone 1 and VL clone 16, as well as the combination clone with both variable domains. (B) Fab fragments of C1, C16, and C1-16 showed improved KD values for muCCL20 when compared with parental AB1. (C) A cell-based β-arrestin assay demonstrated that the affinity-matured variants led to improved CCR6 receptor blocking activity when compared with the parental AB1 clone.
为了评估这些适度的亲和力改善的影响,我们进行了CCR6中国仓鼠卵巢(CHO)-K1β-arrestinG蛋白偶联受体功能测定。 该测定法可以直接测量muCCL20对muCCR6的激活,以及抑制剂对该途径的抑制作用。 这些AB1变体的特征揭示了抗体IC50被AB1-C1,AB1-C16和AB1-C1-16转移了。 尽管AB1-C16的IC50降低了约2倍(即抑制能力提高了2倍),但AB1-C1和AB1-C1-16的IC50降低了约4倍。 这证明了这两个氨基酸改变所赋予的改善的亲和力导致对配体-受体相互作用的抑制作用增强。
2.4 亲和力改善突变的结构见解
分子力学(Molecular mechanics)技术允许在蛋白质系统中进行数千个突变,但缺乏构象采样。 要了解为什么AB1-C1和AB1-C16突变会提高亲和力,我们采用FEP结合增强采样方法与溶质回火( solute tempering )(REST)FEP +进行副本交换[35],以计算增强构象采样框架中的自由能变化。
单突变AB1-C1和AB1-C16均显示出对muCCL20抗原的亲和力提高,分别为–1.00±0.26 kcal mol-1(H:S28R)和–0.16±0.06 kcal mol-1(L:G35Y)。 。 该结果与在结合ELISA中观察到的AB1-C1突变相比于AB1-C16突变更大的亲和力改善相呼应。 对两个代表性突变体结构的检查均显示出显着的重排(图8A和8B),其中在最小化的输入结构与H:S28R和L:G35Y突变体之间分别观察到2.6 A和2.4 A的“主干”(backbone) RMSD。

H:S28R的结构分析表明,在H:28位置引入精氨酸有助于抗原的H:R28和A:Y4之间形成π阳离子相互作用(图8F)。与对接结构一样(图8C),对天然结构的增强采样(图8E)表明L:S28与muCCL20之间不存在相互作用;只有一个结构才对接。因此观察到的自由能变化可以归因于新的分子间相互作用的引入。有趣的是,L:G35突变为酪氨酸也使muCCL20的L:Y35和R66之间引入了新的π阳离子相互作用。相邻的L:D34残基和A:R25之间存在盐桥(图8H),这在对接构象中不存在,可以通过代表性构象来解释,即,人口最多的簇的质心,原始状态。 L:D34 / A:R25相互作用在原始状态下的存在(图8I)表明,自由能的变化不能归因于新盐桥的形成,因为它既存在于参考(天然)中也存在于最终盐中状态。因此,适度的结合自由能变化可以通过π-阳离子相互作用来解释。与H:S28R相比,L:G35Y观察到的ΔΔGbinding的变化较弱,这可能是由于相互作用几何形状较差,为5.7Å/51.5̊,而对于H:R28 / A:Y4则为4.9 A /28.5̊。
尽管这两个突变都是根据MMΔΔE结合计算(H:S28R,–9.07 AU; L:G35Y,–1.46 AU)选择的,但是无法通过将突变映射到对接的构象上来阐明这些相互作用(8C和8G)或通过分子力学方法(图8D和8H),只有使用增强的采样技术才能访问。
三、讨论
在计算机上,亲和力成熟是蛋白质工程和抗体治疗领域一个充满挑战但又很有希望的领域。在这项研究中,我们选择了前所未有的方案,即在低纳摩尔范围内野生型抗体的起始KD不能提供抗体-抗原复合物的共晶体结构,以测试基于计算的亲和力的新潜力成熟。在此案例研究中,仔细考虑了过程中的每个步骤,尤其是蛋白质对接及其所带来的挑战。我们选择使用计算机模拟和实验丙氨酸扫描诱变来确认对接姿势,以建立对预测的信心。有趣的是,在进行丙氨酸扫描预测的情况下,14/23突变显示出结合力的降低,但是当进行实验测试时,仍然有9个突变保持了结合力。这使我们能够在正确的抗体-抗原界面上进行磨合,以设计亲和力改善的突变。考虑到同源性建模,对接和诱变带来的潜在错误的组合,值得注意的是,仅从19种变体中(选自三种不同的突变方案和能量功能),我们就能鉴定出两个亲和力改善突变。
在本研究中证明采用三种协议(Discovery Studios,SchroMMdinger(MM-GBSA)和Rosetta(MM-LKSM))进行亲和力成熟是有用的,因为识别出的两个匹配都没有通过一种以上的方法来拾取(表3)。 AB1-C1(H:S28R)是从Schrödinger协议中发现的,而AB1-C16(L:G35Y)是从Discovery Studio中发现的。与抗体和蛋白质治疗的辅助设计(ADAPT)方法[11]不同,后者使用多种协议来找到方法间的共识,从而增强了对特定突变的信心,我们发现多种方法提供了更大的突变空间和尽管数据集相对有限,但避免了特定的残基或位点偏倚。尽管存在上述挑战,但是MM方法在这项工作中对过滤对接姿势和识别关键残差特别有用。确实,具有实验验证性的丙氨酸扫描的新用途以及可证明的进行重新对接的必要性是这项工作的重要发现。但是,预计在计算丙氨酸扫描中比在计算亲和力成熟中更可靠,因为工程化特定的高能相互作用(如盐桥)的损失比引入新的功能性突变更容易可能需要进行重大的结构重组。
此处概述的具有计算机亲和力成熟度的方法具有潜在的优势,即可以节省大量时间和资源来识别潜在命中。 根据我们的经验,典型的经验性亲和力优化活动可能至少需要6个月的时间,而计算策略与重点实验评估相结合可以节省大量时间。 另外,不需要生成抗原-抗体共晶体结构,我们的方法从典型的基于晶体结构的计算机模拟亲和力成熟工作流程中节省了大约3–18个月的时间。 另外,计算机模拟设计具有将特异性和可开发性设计参数简化为亲和力成熟分子的潜力。
尽管我们的工作强调了将计算方法与实验方法相结合的协同作用,但我们也承认在该领域需要进行重大开发。常规的实验随机诱变策略(例如简约诱变或展示文库方法),有时在共晶体结构的帮助下,对提高抗体亲和力非常有效,通常可以达到10到100倍以上[3,5 ,36]。通过基于晶体结构的集中计算机设计方法来实现该目标更具挑战性[9,37–40],而在缺乏抗体-抗原复合物晶体结构的情况下,成功率甚至更低。最近发表的案例研究表明,单域内抗体与α-突触核蛋白的结合亲和力从微摩尔亲和力提高到66 nM [41]。这项工作和我们的工作已显示出从头抗体亲和力成熟空间发展的令人鼓舞的迹象,但也显示了推动亚纳摩尔亲和力的挑战。另外,有机会从19个突变体中的2个提高阳性命中率,这与在随机诱变方法中观察到的相差不大。我们已经表明,诸如FEP +之类的方法可以提供一些进展。但是,该方法尚未针对生物制品的应用进行优化,只能准确预测单点突变的影响。考虑到蛋白质动力学,侧链灵活性和基于物理学的自由能计算,用作饱和诱变方法在计算上也将花费巨大。利用多种MM-GBSA / MM-LKSM方法也可能在这方面提供进步的机会,作为伪采样方法,其中每种方法的能量函数以独特的方式对构象进行采样,并且可能揭示出被忽视的影响通过其他协议。
在这里,我们介绍了在没有共晶结构的情况下进行计算机亲和力优化的有趣案例研究。 我们展示了一种新颖的策略,可使用计算机模拟预测和实验指导的集成方法来实现适度的亲和力改善。 尽管我们的工作强调了技术状态的局限性,例如对大型蛋白质复合物需要更准确的能量预测方法,但我们希望为计算蛋白质设计和工程领域的发展提供一条途径。
四、材料和方法
4.1 抗体同源性建模
AB1 Fv模型于2017年2月15日用SAbPred [29]生成,使用Abody-Builder [42]从框架化的抗体结构数据库中搜索框架模板结构,在本例中为VH和VL结构域搜索1MH5 。 SabDab [43]和CDR环用FREAD [44]建模。
4.2 抗体-抗原对接
muCCL20的核磁共振溶液结构是从蛋白质数据库(PDB ID,1HA6)获得的[45]。该结构集合由20个低能结构组成,其中第一个代表该集合的平均结构,并用于随后的计算机模拟实验。
将抗体Fv和muCCL20结构导入到Discovery Studio 2016软件包[32]中,并用清洁蛋白工具制备两个结构,然后在对接之前用CHARMM-Polar-H力场[46]进行输入。用ZDOCK [47]进行刚体对接,使用的步距大小为15°,距离截止值为10Å,并且抗体的非IMGT CDR残基被封闭。随后的改进是用RDock进行的[47]。
在进行丙氨酸扫描诱变和结合研究后,抗体和抗原被重新对接。采用了相同的方案,但附加的参数是,被认为对结合至关重要的所有六个AB1残基必须在muCCL20的10Å之内。
4.3 计算诱变 In silico mutagenesis
首先对五个最佳候选姿势中的每个姿势进行计算机丙氨酸扫描。 在Discovery Studio 2016软件包中进行诱变,并在计算ΔΔE之前将每个突变体的结构最小化。 还使用相同的Discovery Studio协议(用于丙氨酸扫描)对重定构象进行诱变,其中对1925个输出姿势中的每个姿势进行10个单点突变。
4.4 三种方法的计算机亲和力成熟
Discovery Studio protocol
在Discovery Studio 2016 GUI中将复杂结构与CHARMM-Polar-H力场一起应用。 然后将类型化的结构导出并用作诱变的基础。 使用针对Discovery Studio的Perl脚本,我们将hybrid CDR残基突变为所有20个遗传编码的氨基酸,并计算了ΔΔEbinding和ΔΔEstability值,这导致1400个突变。
Schrodinger biologics suite protocol
使用Schrorodinger Biologics Suite 2016-3 [33]中的蛋白质制备向导工具(Protein Preparation Wizard tool)[48,49]可以制备复杂的结构。 通过使用PropKa [50,51]根据其环境对可滴定的(titratable)残基进行质子化,并使用OPLS3 [52]力场对其进行分型,然后将结构最小化。
由于优化更广泛的突变环境可能会导致不必要地接受大侧链,因此进行了两个单独的诱变实验。 一个实验使用了0.0Å的最优化距离,即仅突变的reside,并且还最小化了5.0Å内的任何残基。 通过同时使用优化标准和接受突变的共识,有可能避免选择会在空间上破坏本地蛋白质环境的残基,同时鉴定出仅需进行很小的结构重排即可进入构象的残基,从而提高结合亲和力。
Rosetta protocol.
在诱变之前,使用Rosetta [34]中的clean_pdb.py工具清洁复杂的结构,并使用Relax协议将空间冲突消除为最小。 将500个最小化的结构按其总分进行排名,并使用具有最低总能量的结构进行诱变。
使用Rosetta脚本协议进行诱变,该协议用于优化每个CDR位置的突变。 在mutagenesis.xml(S1文本)中,ΔΔEstability过滤器设置为0.0,因此仅接受不会破坏复合物或界面不稳定的突变。 为每个位点生成一百个结构,并通过从score.sc文件中提取最佳分数并将相应的PDB与天然蛋白质进行比较,从而鉴定出最稳定的突变。 除了“结果”部分所述的ΔΔE结合计算(请参阅“计算机亲和力成熟度”)之外,还使用Rosetta pmut(点突变)方案来预测所有突变的ΔΔEstability值。
与WaterMap进行突变比较。
使用SchrödingerBiologics Suite 2017-4对母体复杂结构进行了WaterMap [53]分析。 87个单突变的结构取自Schrodinger 5.0 A细化方案,并映射到WaterMap结果中。 目视检查以确定每个突变体是否都可以置换一个或多个WaterMap水分子。 如果这样,则将每个置换的分子的ΔG值合并。
改进的亲和力突变的结构合理化。 Structural rationalisation of improved affinity mutations.
FEP +是FEP [15]和增强采样方法REST [54-56]的组合,除了双重突变外,还用于两个亲和力改善突变的后合理化。 FEP和REST的组合使系统能够在突变时探索更多的构象状态,并且应该提供对自由能和结构变化的更可靠的估计。
FEP +计算在Schrorodinger Biologics Suite 2017-4 [57]中进行,使用20 ns的仿真时间来确保自由能收敛。
4.5 Mutant panel construction, expression, and purification.
略
4.6 Binding ELISA
略
4.7 Fab binding affinity surface plasmon resonance measurements.
略
4.8 In vitro muCCL20 activity.
参考资料
- Cannon DA, Shan L, Du Q, Shirinian L, Rickert KW, Rosenthal KL, et al. (2019) Experimentally guided computational antibody affinity maturation with de novo docking, modelling and rational design. PLoS Comput Biol 15(5): e1006980. https://doi.org/10.1371/journal. pcbi.1006980
