【4.3.7】基于序列或结构预测非连续BCE--ElliPro
网址:http://tools.iedb.org/ellipro/
摘要
抗体或B细胞表位的可靠预测仍然具有挑战性,但对于疫苗设计和免疫诊断而言却非常需要。 证明了抗原性,溶剂可及性和蛋白质柔韧性之间的相关性。 随后,Thornton及其同事提出了一种方法,用于鉴定从蛋白质球状表面突出的蛋白质区域中的连续表位。 这项工作的目的是将该方法作为网络工具实施,并评估其在从抗体-蛋白质复合物结构已知的不连续表位上的性能。
在这里,我们介绍ElliPro,这是一个实现Thornton方法的网络工具,它与残基聚类算法,MODELLER程序和Jmol Viewer一起,可以预测和可视化给定蛋白质序列或结构中的抗体表位。 ElliPro已在从抗体-蛋白质复合物的3D结构推断出的不连续表位的基准数据集中进行了测试。 与其他六种可用于表位预测的基于结构的方法相比,当考虑到每种蛋白质的最重要预测值时,ElliPro表现最佳,AUC值为0.732。 由于对70%以上的蛋白质而言,最佳预测的排名最多是前三名,并且从未超过五个,因此ElliPro被认为是鉴定蛋白质抗原中抗体表位的有用研究工具。 可在 http://tools.immuneepitope.org/tools/ElliPro 上找到ElliPro。
ElliPro的结果表明,考虑到更多区分抗原表位与非表位的特征,对抗体抗原表位的进一步研究可能会进一步改善预测。 由于ElliPro基于蛋白质结构的几何特性,并且不需要训练,因此它可能更广泛地用于预测不同类型的蛋白质-蛋白质相互作用。
一、背景
抗体表位,也称为B细胞表位或抗原决定簇,是被免疫系统的特定抗体分子或特定B细胞受体识别的抗原的一部分[1]。对于蛋白质抗原,表位可以是蛋白质序列中的短肽,称为连续表位,也可以是蛋白质表面上的原子斑(patch of atoms),称为不连续表位。尽管连续表位可直接用于疫苗和免疫诊断的设计,但不连续表位预测的目的是设计一种分子,该分子可模仿表位的结构和免疫原性,并在抗体生产过程中替代它。在医学诊断或实验研究中,可以将表位模拟物视为预防性或治疗性疫苗或抗体的检测方法[2,3]。
如果使用可用的免疫原性肽集合构建的依赖序列的方法可以预测连续表位(综述见[4]),则不连续表位-大多数情况是免疫蛋白识别完整蛋白质,病原病毒或细菌时系统–在不了解蛋白质的三维(3D)结构的情况下,很难从功能测定中预测或鉴定[5,6]。基于3D蛋白质结构的表位预测的首次尝试始于1984年,当时晶体学温度因子与烟草花叶病毒蛋白,肌红蛋白和溶菌酶的几种已知连续表位之间建立了相关性[7]。还发现抗原性,溶剂可及性和蛋白质中抗原性区域之间的相关性[8]。 Thornton及其同事[9]提出了一种鉴定从蛋白质球状表面突出的蛋白质区域中连续表位的方法。高突出指数值(high protrusion index )的区域显示出与实验确定的肌红蛋白,溶菌酶和肌红蛋白的连续表位相对应[9]。
在这里,我们介绍ElliPro(源自Elli psoid和Pro trusion),该网络工具实现了Thornton方法的修改版本[9],并与残基聚类算法,MODELLER程序[10]和Jmol浏览器一起,允许 蛋白质序列和结构中抗体表位的预测和可视化。 ElliPro已在从抗体-蛋白质复合物的3D结构推断出的抗原决定基的基准数据集上进行了测试[11],并与六种基于结构的方法进行了比较,包括仅有的两种专门为抗原决定簇预测而开发的现有方法CEP [12]和DiscoTope [ 13]; 两种蛋白质-蛋白质对接方法,DOT [14]和PatchDock [15]; 以及两种基于结构的蛋白质-蛋白质结合位点预测方法,PPI-PRED [16]和ProMate [17]。 可在 http://tools.immuneepitope.org/tools/ElliPro 上找到ElliPro。
二、应用 Implementation
2.1 工具输入
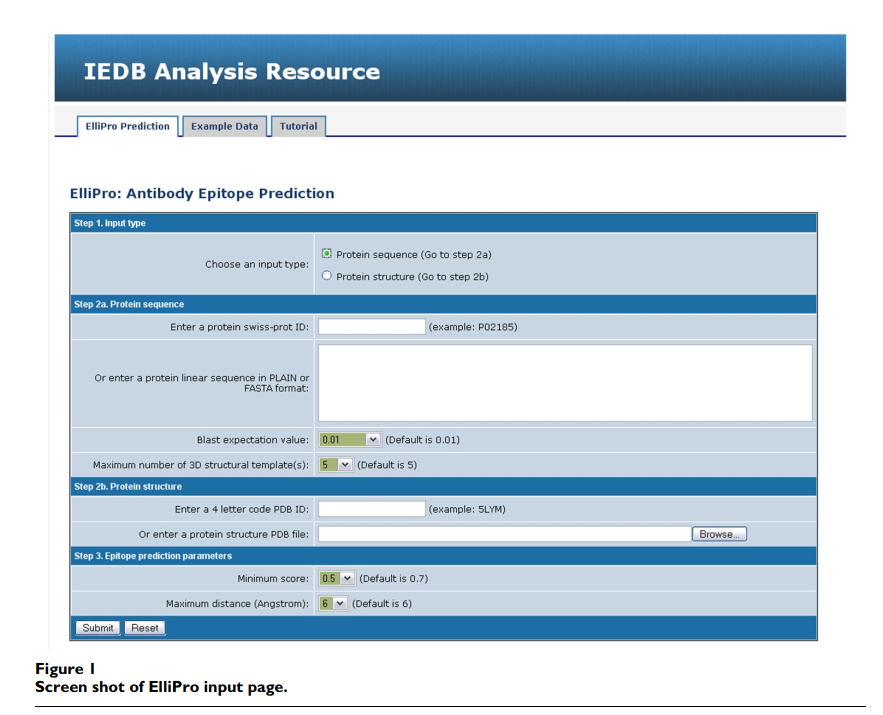
ElliPro被实现为可通过Web访问的应用程序,并接受两种类型的输入数据:蛋白质序列或结构(图1,步骤1)。在第一种情况下,用户可以输入蛋白质SwissProt / UniProt ID或FASTA格式或单个字母代码的序列,并选择BLAST e值的阈值和PDB中将用于建模的结构模板的数量提交序列的3D结构(图1,步骤2a)。在第二种情况下,用户可以输入四个字符的PDB ID或以PDB格式提交自己的PDB文件(图1,步骤2b)。如果提交的结构包含一个以上的蛋白质链,ElliPro将要求用户选择计算所基于的链。用户可以更改ElliPro用于表位预测的参数的阈值,即,最小残基得分(突出指数)(在此表示为S)在0.5到1.0之间,最大距离在R中表示为4 – 8Å。
2.2 3D结构建模
如果使用蛋白质序列作为输入,ElliPro会使用BLAST搜索[19]在PDB中搜索蛋白质或其同源物[18]。 如果在PDB中找不到符合BLAST标准的蛋白质,则运行MODELLER [10]来预测蛋白质3D结构。 用户可以更改BLAST e值的阈值和MODELLER用作输入的模板数量(图1,步骤2a)。
2.3 ElliPro方法
ElliPro实现三种算法来执行以下任务:
- 近似蛋白质形状的椭球[20];
- 残基突出指数(PI)的计算[9];
- 基于相邻残基的PI值进行聚类。
Thornton的连续表位预测方法基于两个首个算法,并且仅考虑了Cα原子[9]。它将蛋白质表面近似为椭圆体,其大小可以变化,以包含不同百分比的蛋白质原子。例如,90%的椭球包含90%的蛋白质原子。对于每个残基,突出指数(protrusion index,PI)定义为包围在椭球中的蛋白质原子的百分比,在残基处蛋白残基首先位于椭球之外;例如,所有90%椭球体之外的残基的PI = 9(在ElliPro中为0.9)。在实现前两个算法时,ElliPro与Thornton方法不同,它考虑了每个残基的质心而不是其Cα原子。
用于对残基进行聚类的第三种算法基于突出指数S的阈值和每个残基质心之间的距离R定义了不连续的表位。计算不连续表位时,应考虑所有PI值大于S的蛋白质残基。将单独的残基聚类到不连续的表位中涉及三个步骤,这些步骤递归地重复进行,直到没有重叠残基的不同簇形成为止。
- 首先,主要簇是由单个残基及其相邻残基在距离R内形成的。
- 第二,次要簇是由主要簇形成的,其中至少三个质心在距离R内。
- 第三,第三类簇由含有共同残基的第二类簇形成。这些残基的第三级簇代表蛋白质中预测的独特的不连续表位。每个表位的得分定义为在表位残基上平均的PI值。
2.4 预测表位的3D可视化
开源分子查看器Jmol [21]用于可视化蛋白质3D结构上的线性和不连续表位。 表位可视化的示例如图2所示。

三、结果和讨论
为了评估ElliPro的性能并与其他方法进行比较,我们使用了先前建立的用于不连续表位的基准方法[11]。 我们在39种蛋白质结构中存在的39种抗原决定簇的数据集上测试了ElliPro,其中基于具有单链蛋白质抗原的两链抗体片段的3D结构,只有一个不连续的抗原决定簇[11]。
根据参数R和S的阈值,ElliPro可以预测每种蛋白质中不同表位的数量。 如果R为6Å,S为0.5,则分析的每种蛋白质的预测表位平均数为4,变异范围为2至8。例如,间日疟原虫钩虫表面蛋白Pvs25 [PDB:1Z3G,链A], ElliPro预测了四个表位,分别为0.763、0.701、0.645和0.508(图2)。
对于每种蛋白质中的每个预测表位,我们计算了正确(correctly,TP)和错误预测的表位残基(FN)和非表位残基,它们被定义为所有其他蛋白质残基(TN和FN)。使用费舍尔精确检验(右尾)确定预测的统计显着性,即预测表位/非表位中实际表位/非表位残基的观察频率与预期频率之间的差异。如果P值= 0.05,则认为该预测很重要。然后,针对每个预测计算以下参数:
-
灵敏度(Sensitivity,recall,召回率或真实阳性率(TPR))= TP /(TP + FN): 正确预测的表位残基(TP)占表位残基总数(TP + FN)的比例。
-
特异性(Specificity, 或1 –假阳性率(FPR))= 1-FP /(TN + FP): 正确预测的非表位残基(TN)相对于非表位残基总数(TN + FP)的比例)。
-
阳性预测值(PPV)(precision,精度)= TP /(TP + FP): 正确预测的表位残基(TP)相对于预测的表位残基总数(TP + FN)的比例。
-
准确度(Accuracy,ACC)=(TP + TN)/(TP + FN + FP + TN)–正确预测的表位和非表位残基相对于所有残基的比例。
ROC曲线下的面积(AUC)–表示TPR对FPR依赖性的图表下的面积; 即对1种特异性的敏感性。 AUC给出了该方法的一般性能,并且“等效于分类器将随机选择的阳性实例的排名高于随机选择的阴性实例的概率” [22]。
例如,对于间日疟原虫钩虫表面蛋白Pvs25 [PDB:1Z3G,链A]中的第一个预测表位,图2, R =6Å,S = 0.5,TP = 13,FP = 13,TN = 156, FN = 4,P值= 5.55E^-10,灵敏度为0.76,特异性为0.92,精度为0.91,AUC为0.84。 补充材料中提供了每个表位的ElliPro性能的结果和详细统计数据,以及R和S的其他阈值[请参见附加文件1]。
表1和图3给出了所有表位的平均统计量,并根据FP,FN,TP和TN值汇总了整个表位和非表位残基的总值,得出了总体统计量。除ElliPro之外的方法结果 如[11]中所述获得。 根据AUC值,ElliPro表现最佳,将每种蛋白质的最高得分预测值考虑时,将得分S设置为0.7,距离R设置为6Å,将得分S设置为0.5且距离R设置为6Å 考虑了最佳的显着性或平均预测。 使用这些阈值描述结果(表1,图3); 其他阈值的结果在补充材料中提供[请参见附加文件1]。
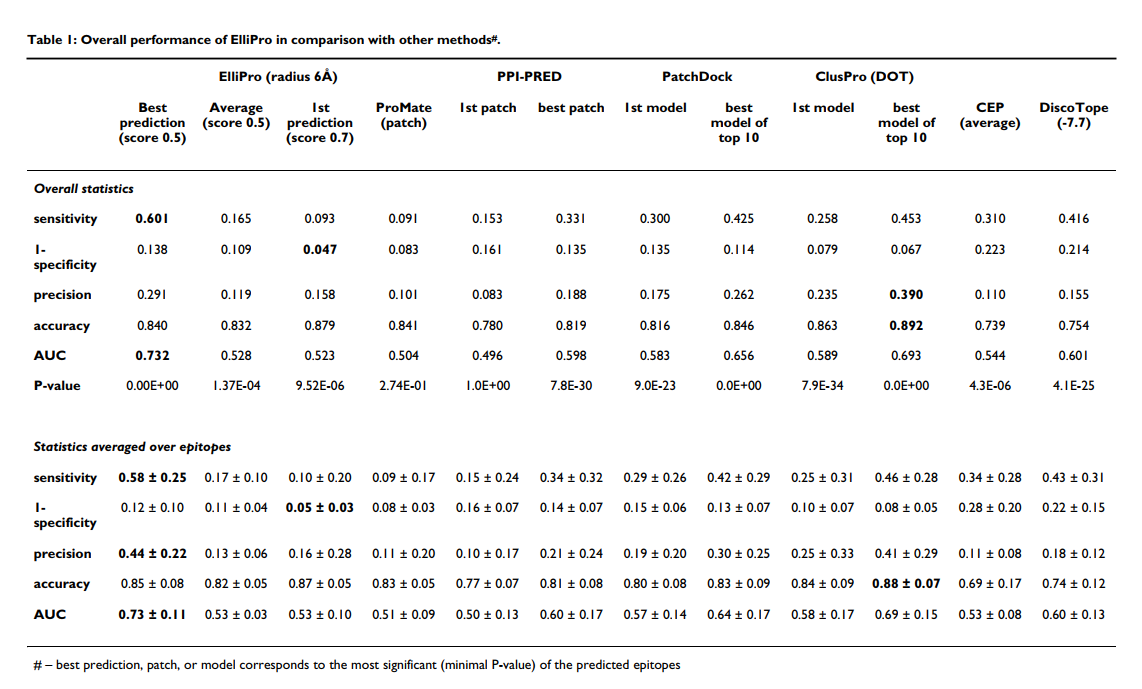
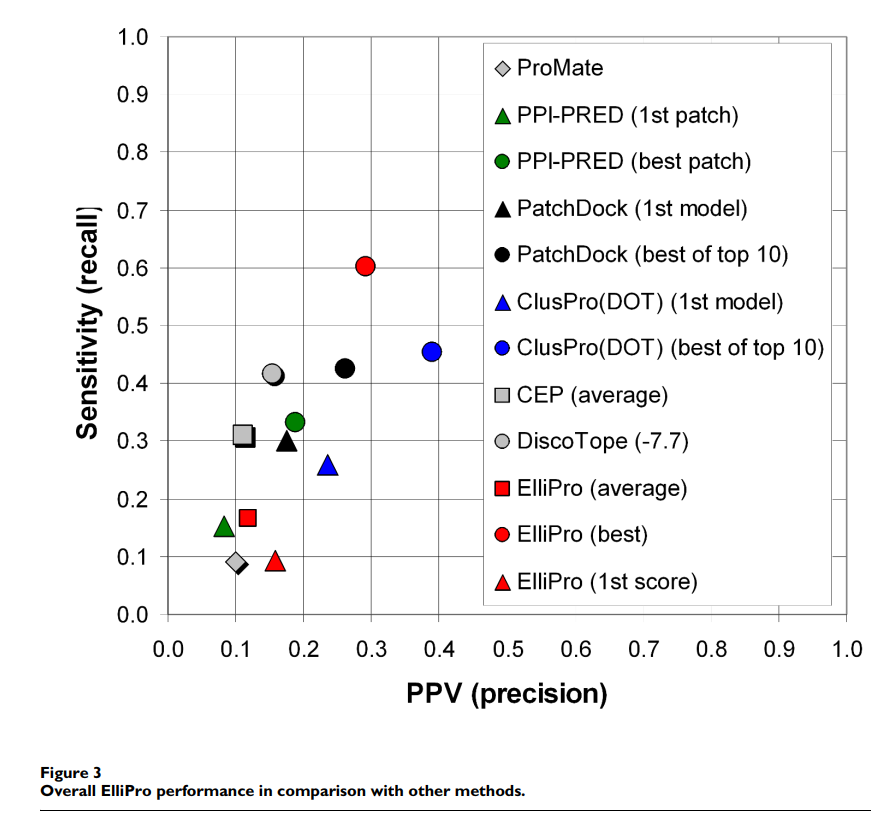
ElliPro的最高预测(即得分最高的预测)与从抗体-蛋白质复合物的3D结构得知的不连续表位相关性较弱(表1,整体统计,AUC = 0.523)。 DiscoTope和对接方法的第一个模型表现更好,AUC值均大于0.6,而蛋白质-蛋白质结合位点预测方法ProMate和PPI-PRED表现较差。但是,当考虑到得分最高的第一个预测时,ElliPro是所有基于特异性的方法中最好的(1-特异性= 0.047),并且在精度上与DiscoTope相当(PPV = 0.158)(表1,总体统计)。
在下一组指标中,当在每种方法的前10个预测中选择最佳匹配时,我们比较了预测方法之间的性能。该方法考虑到每种抗原具有针对不同抗体的多个不同的结合位点。因此,可以预期,最高预测位点不一定会被数据集中使用的特异性抗体识别。此比较仅直接适用于DOT和PatchDock以及ElliPro的对接方法。对于DiscoTope,仅预测一个表位,而对于CEP,没有排名可用于识别前10个预测。
与ElliPro相比,DOT和PatchDock的对接方法具有固有优势,因为它们使用蛋白质抗原和来自同一抗体-蛋白质复合物的抗体的结构来预测结合位点。令我们惊讶的是,当考虑到每种蛋白质的最佳显着预测值时,在所有比较方法中,ElliPro的AUC值最高,为0.732,最高灵敏度为0.601,第二精度为0.29(表1;图3)。 ,红色圆圈)。对接方法给出的DOT的AUC值为0.693,PatchDock的AUC值为0.656,同时还考虑了前十名的最佳预测(表1,总体统计数据;图3)。分析的蛋白质的预测表位的平均数为4,最佳预测值的排名最高为第五。对于一半以上的蛋白质,排在第一或第二位,对于所有蛋白质的70%以上,排在第一,第二或第三位[请参见附加文件1]。
ElliPro基于简单的概念。
- 首先,从蛋白质球状表面突出的区域更易于与抗体相互作用[9]2. 其次,可以通过将蛋白质视为简单的椭球体来确定那些突出[20]。
显然,并非总是如此,尤其是对于多域或大型单域蛋白。但是,未发现蛋白质大小(ElliPro性能)之间的相关性(ElliPro性能介于51到429个残基之间,平均值为171)或结构域数目(分析的39个中的8种蛋白质包含一个以上结构域)(数据未显示) 。
四、结论
ElliPro是基于网络的工具,用于预测给定序列或结构的蛋白质抗原中的抗体表位。它实现了以前开发的方法,该方法将蛋白质结构表示为椭球,并计算了椭球外部蛋白质残基的突出指数。 ElliPro在从抗体-蛋白质复合物的3D结构推断出的不连续表位的基准数据集中进行了测试。与可用于表位预测的其他六种基于结构的方法相比,当考虑到每种蛋白质的最重要预测时,ElliPro表现最佳(AUC值为0.732)。由于在70%以上的蛋白质中,最佳预测的排名最多为3个,并且从未超过5个,因此ElliPro被认为是鉴定蛋白质抗原中抗体表位的潜在有用研究工具。
虽然ElliPro已在抗体-蛋白质结合位点上进行了测试,但可能会在其他蛋白质-蛋白质相互作用上对其进行测试,因为它实施了一种基于蛋白质结构的几何特性且不需要培训的方法,可能会很有趣。
与基于训练并利用表位特征(例如氨基酸倾向,残基溶剂可及性,空间分布和分子间接触)的DiscoTope进行比较,表明对抗体表位的进一步研究考虑了更多区分表位与非表位的特征 ,可以改善抗体表位的预测。
参考资料
- Ponomarenko, J., Bui, H., Li, W. et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics 9, 514 (2008). https://doi.org/10.1186/1471-2105-9-514. 网址:https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-9-514
