【1】计算在生物药物开发中的应用
一、介绍 INTRODUCTION
生物制剂,特别是单克隆抗体和基于抗体的疗法(抗原结合片段[Fabs],抗体-药物偶联物[ADC]和片段可结晶[Fc]融合蛋白)已经成为一种重要的治疗学家。过去几十年。根据日本mAb提供的信息,截至2014年2月,美国(US)和欧盟(EU)批准了30多种基于抗体的治疗药物,用于预防或治疗多种人类疾病,包括几种癌症,呼吸道合胞病毒(RSV)感染,类风湿性关节炎,牛皮癣,哮喘,黄斑变性,多发性硬化,骨质流失和系统性红斑狼疮。除此之外,美国,欧盟或日本的监管机构目前正在审查另外10种基于抗体的治疗方法。根据最近的预测,2014年,目前处于第3阶段临床试验的4或5种治疗性单克隆抗体(mAb)候选物预计将递交监管审查,预计另有3或4种分子将被批准在美国进行营销或欧盟。因此,预计在不久的将来可用于治疗人类疾病的抗体疗法的强劲增长。基于抗体的疗法在诊所中的成功正在通过生物学,抗体设计,制造(manufacturing),分析表征,制剂开发和这些候选物的递送装置(delivery devices)的等若干技术飞跃而得以实现。
Availability of affordable, safe, and easily deliverable biologics can signi cantly expand the reach of biologics to all parts of the world and improve health for all humans
近年来,这些生物技术进步正以越来越快的速度发展,并促进了创新者和后续生物制剂的发展。生物制剂在诊所的成功也推动了全世界对这些药物的需求。然而,这些生物技术的成功也伴随着药物开发成本,监管障碍以及临床前和临床阶段治疗候选药物失败率的急剧增加。在实验室中发现的治疗候选人转化为临床可用药物的比率正在下降。总体而言,高药物开发成本和药物制造商的预期导致高价格制度,付款人(政府,保险公司和个体患者)越来越难以负担。因此,目前生物药物发现和开发的实践需要创新,以降低药物开发成本,提高安全性和灵活,友好的交付选择。提供价格合理,安全且易于交付的生物制剂可以显着扩大生物制剂在世界各地的影响范围,并改善所有人类的健康状况
本章以及本书的目的是强调计算工具和分析如何有助于生物药物的发现和开发,并描述一种称为生物制药信息学(biopharmaceutical informatics)的新学科。与小分子药物不同,生物制剂的发现和开发要经过一系列试验和错误。然而,目前可用的计算工具可用于药物发现和开发周期的每个阶段,从目标验证到先导化合物的设计/选择,到生物过程优化和配方开发,再到安全性,有效性和药代动力学,再到临床试验设计。例如,分子设计/选择和生物过程开发可以显着影响生物制药产品的生物活性和安全性。因此,对物理化学降解,热力学稳定性,免疫原性的生物候选物进行严格的分子水平评估,在候选人设计和选择的早期阶段,其他药物安全属性可以非常有助于预测开发它们所需的资源并防止晚期药物失效。在早期阶段,优化主要候选物的氨基酸序列以获得比仅针对靶标结合性优化的候选物更容易,成本有效的药物开发和安全性方案也是可行的。
生物制药信息学致力于使用信息技术,基于序列和结构的生物信息学分析,分子建模和模拟,以及针对生物药物开发的统计数据分析。在本章中,我们将研究生物制药信息学在生物药物开发中的几种应用。我们专注于了解基于抗体的生物制剂的物理化学降解的潜在分子起源以及这些可能与产品质量和安全性有关。开发包含生物物理稳定性,安全性和临床前/临床免疫观察的实验数据的数据库,以及一些生物候选物的分子序列结构分析,可以将表现良好的候选物与表现不良的候选物在分子水平特性与那些候选物进行比较。 这种比较将提高我们对生物候选物的分子水平特性如何影响其作为药物产品的发展的理解。在这里,我们通过自己的工作展示案例研究。本章的后续章节将更详细地描述本章的几个方面。因此,本章不全面回顾文献。相反,我们的目标是通过突出有趣的科学进展和案例研究,激发读者对计算应用的不同方面对生物制药药物开发的兴趣。这里有必要提及的是,计算也越来越多地应用于药物发现期间生物候选物的基于结构的设计,以改善这些分子对其同源靶标的结合亲和力和选择性。这是生物制药信息学的一个重要方面。但是,这超出了本章的范围。在生物制药信息学的另一个方面,计算工具也通过系统药理学和药代动力学/药效学建模应用于药物开发。这些领域也未在本章中介绍。
二、信息技术应用:信息供应链(information supply chain)与基于知识的决策(knowledge-based decision making)
从本质上讲,生物药物的开发是一项信息业务。除了物理临床用品外,所有开发产品实际上都是信息,满足了初级(预期)和二级(非预期)客户对所有利益相关者开发和许可高价值产品的需求。 无论是验证分析方法,强大的制造过程理解,适合用途的配方设计,质量评估,临床供应管理,监管互动,预测模拟等,都必须开发和维护有效的信息供应链,满足研发内部和外部客户的需求。因此,必须始终关注实现基于知识的方法,通过在整个开发价值流中轻松访问高质量数据,促进和承保高质量的决策,开发和执行。要实现这一目标,必须经济有效地捕获(生产),管理(策划)并以维持和利用我们的集体知识遗产的方式提供(分发)所有相关信息。从本质上讲,有效的信息供应链体现为一种基于知识的开发引擎,在这种引擎中,合适的人员可以在合适的时间以正确的格式访问必要的信息/知识,从而在整个开发周期中实现价值创造。
采用基于知识的方法的愿望很容易陈述,但在操作细节中难以描述,因为它包含了人类的意图和文化以及工具和技术。 理解和利用技术实现共同目标的意愿同样重要,使其成为技术和人类企业。 同样重要的是要注意,知识管理( knowledge management)不是组织构建或购买的东西。 相反,它仅仅是开发一种专用信息供应链的结果,该供应链将适当的技术嵌入在知识增值和意识文化中运作的有效工作流程中。 这三者对成功至关重要,但趋势是仅关注技术。
在剖析有效信息供应链(生产,分销,消费)的主要要素之前,我们必须认识到两个基本信息市场,因为它们对设计和文化都有独特的影响。主要数据市场是最熟悉的一个,其中请求生成信息的客户也是功能封闭的请求 - 交付循环中的客户,例如流程开发,使用特定标准和工作请求分析开发的样本分析。这些交易很好地描述,可重复,并且易于嵌入到工具和流程中。预先商定数据标准,质量,格式和报告。正在测试的假设,例如产品质量与规格,也很合理。另一方面,二级数据市场是信息消费者没有要求最初创建信息,而是寻求从其他原因创建的现有信息中提取额外价值的市场,例如,将临床事件数据与批次相关联处置/特征数据以建立关键质量属性。这些信息查询的独特之处在于,数据从未被收集或策划,最初是为了测试这样的假设本身,因此,当信息出现时,数据捕获,管理和报告的要求并不是为此目的而定义的。创建。从历史上看,重点一直放在主要数据市场上,对二级数据市场几乎没有文化方面的考虑。这种文化观点是可以理解的,但它阻碍了我们从生物药物开发数据中提取全部价值的能力。生物制药信息学的发展需要更广泛的学习观点,使假设生成,而不仅仅是假设检验
从最简单的意义上讲,信息供应链(information supply chain)包括数据捕获(生产),数据管理(库存管理)和报告(分销渠道)。 在大多数主要数据市场中,所有三个要素都包含在平台工具中,如实验室信息系统(LIMs),电子Labortatory Notebook(eLN)和数据库结构。 这些包括数据创作,数据存档,检索和报告,所有这些都在生产者和消费者共同的工具中,符合良好的标准。 在二级数据市场中,生产者和消费者通常不共享专业知识或访问共同工具或共享数据标准或定义的标准,使得相关信息的暴露和检索即使不是不可能也是困难的。
因此,一个有效的信息供应链,忠实地服务于无数生物药物开发利益和合作伙伴的主要和次要数据市场,必须具备以下能力:
- 认识到所有数据都具有遗产和特定价值,并将其视为具体价值的一种文化
- 能够捕获,策划和使用其生成的信息的文化
- 有利于数据检索和重用的常用数据标准和质量标准
- 有效的工具互操作性,可实现数据发现和分发
- 工作流程一致性促进最终用户以适当格式分发数据
- 信息整合层功能,使不同的系统在必要或更实用时可以作为一个系统工作
- 适当的数据分析,以减少和提取我们的信息含义
要获得这些功能,通常需要进行以下投资(investments):
- 用于暴露和检索所需信息的工具,无需依赖社交网络,当地专家或对多个策展点的深入了解
- 在工作流和合作伙伴之间捕获适当的元数据,并提供足够的上下文,以促进有意义的解释
- 开发直观的最终用户界面,以最少的培训或特定的工具专业知识促进遵守业务规则
- 项目组合管理系统,可实现快速,明智的决策制定,并保留跨项目的遗留学习
- 实施关键信息的单一来源,以减少冗余,低效率和交叉验证负担
- 信息利用率完全超出书面文字 - 音频,视频和图像
- 访问与技术无关的信息的工具
上述讨论提出了一个有利的路线图,用于利用信息技术实现生物制药信息学的目标。 除此之外,还需要开发科学理解,工具和技术,以充分发挥这一领域的潜力。 本章后面的部分描述了我们最初的尝试。
三、生物候选物的可开发性评估:预测可能的物理化学降解位点
传统上,生物药物的发现和开发已被划分为发现和开发部门。在药物发现期间,主要目标是鉴定最有可能实现期望治疗效果的高效候选分子。在这个阶段,高特异性受体的亲和性和选择性是分子候选物设计/选择的主要驱动因素。一旦被选中,生物候选物就会进行开发,进行动物和人体测试的各个阶段的安全性和有效性。药物开发科学家致力于稳定生物分子,以实现商业上可行的生产,长保质期和以用户友好的形式交付。这种传统方法意味着所选分子的氨基酸序列是固定的,并且一旦进入发育阶段就不能改变。因此,药物产品开发主要涉及使用外部工艺,如配方缓冲液,pH和赋形剂筛选,冻干和药物递送装置,尽量减少药物分子在储存,运输和给药期间的物理化学降解。这种范例的局限性在于,如果最佳配方和递送组合证明是困难的,则药物产品开发失败或停滞在具有差的稳定性或溶解性的有问题的分子上。即使最佳生物制药药物产品是由具有较差生物物理属性的分子开发的,其递送选择也可能是有限的。例如,开发用于肠胃外的生物药物产品可能是可行的,但由于高浓度下的粘度和可注射性相关问题,因此不能用于皮下给药。该范例的另一个限制是优化所选择的候选物质以获得所需属性的机会,例如高细胞系表达产率,安全性(低免疫原性),改善的药代动力学/药效学(PK / PD),较低频率的给药和灵活的递送选择。此外,缺乏对发现和开发工作的考虑,导致昂贵的开发,即使不是完全失败。所有这些限制都导致了目前开发和生产生物制剂的高成本。为了克服上述限制并利用生物药物产品开发计划可用的所有机会,必须修改上述区域化范例。这可以通过了解生物候选药物的序列和结构特征并优化它们来实现,不仅是效力,还有可开发性,制造成本和安全性。下面,我们通过考虑潜在的化学降解位点,聚集,免疫原性和高浓度溶液行为,描述如何使用计算来评估生物学候选人的可开发性。
生物制剂包括多种产品,如寡核苷酸,生长因子,细胞因子,激素,受体,酶和凝血因子,预防/治疗疫苗,单克隆抗体(mAb),抗体成分(Fabs),Fc融合蛋白和抗体-药物结合物(ADC)。 这些大分子具有高度复杂的异质三维(3D)分子结构,并且使用重组DNA技术在多种宿主中产生或可以是血浆衍生的。 图1.1通过比较mAb与小分子药物的分子结构,说明了生物药物分子的复杂性。 如前所述,单克隆抗体正在成为最成功的生物制药类别,可以看出它们的分子结构比小分子药物更大,更复杂。 基于单克隆抗体和抗体的候选药物和产品是本章的重点。
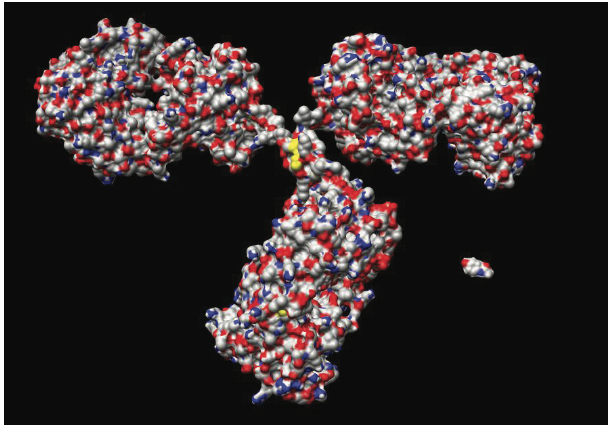
图1.1显示了mAb与小分子药物的分子结构。 在这种情况下显示的mAb是鼠IgG2a mAb18,其晶体结构可在Protein Data Bank entry 1IGT中获得。 这里显示的小分子药物是对乙酰氨基酚。
由于它们的尺寸和结构复杂性,生物制剂在制造,运输,储存和管理过程中易受许多物理化学应力的影响。由这些压力引起的降解可能会损害这些药物产品的效力和安全性。生物制剂可能遇到的许多物理化学应力如图1.2所示。这一点说明生物分子在不同阶段所经历的多重胁迫可导致共同的物理退化,例如聚集。在适应极端环境条件(如高温和低温,高酸度和高盐度)的生物体中发现的天然蛋白质也面临着相似的压力。因此,自然界使用的策略可能会应用于生物制剂的分子设计和配方。特别是,适应高温和低温的生物(嗜热生物和嗜冷菌)可以教会我们在提高蛋白质稳定性和溶解度方面的重要教训,而不会产生牺牲效力或导致大规模的结构重排。在蛋白质活性,稳定性,溶解度和粘度之间取得平衡与生物逻辑产品开发的目标一致,即保持蛋白质的分子(物理化学和结构)完整性以应对环境压力。
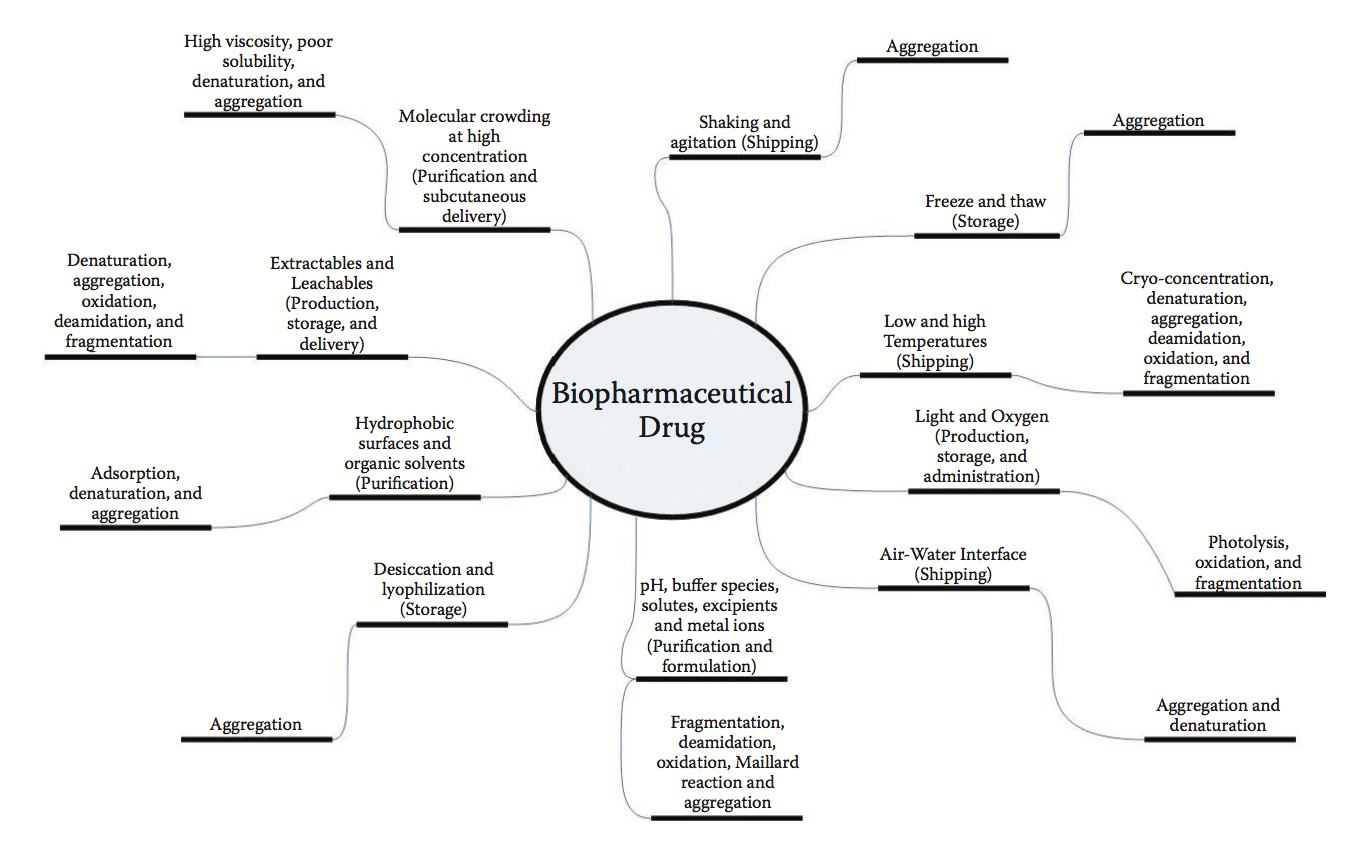
图1.2 生物制药药物在生产,运输,储存和管理的各个阶段面临的物理化学应力(棒的内圆)及其后果(棒的外圆)。 请注意,药物制造,储存和运输的不同阶段可能会遇到常见的压力。
在活细胞中,由于富含氧的水环境,蛋白质会因几种非酶促共价修饰而逐渐老化。此外,天然蛋白质中发现的几乎所有氨基酸残基都易受一种或另一种化学降解的影响。同样,可以想象生物制剂也可以在储存期间老化。因此,生物制剂必须含有阻碍药物物理化学降解的成分,并在其保质期内保持生物药品的完整性。已经描述了生物制药的几种潜在的物理化学降解及其机制。其中,脱酰胺,氧化,异构化(isomerization),断裂和聚集是常见的。
大多数化学降解途径(即脱酰胺作用,氧化作用等)来自蛋白质序列中发现的特定残基或残基对。这些降解位点可以容易地在生物候选物的氨基酸序列中鉴定。易受β-链介导的聚集影响的聚集倾向区域(Aggregation-prone region,APR)通常长5-10个残基,并且也可以使用氨基酸序列预测。然而,APR预测是复杂的,并且当前可用的方法不是100%准确的。此外,β-链介导的聚集可以导致几种形态,范围从无定形β-聚集体到淀粉样蛋白膜。使用实验数据可以得到形成无定形β-聚集体或淀粉样蛋白的六肽。Thangakani等开发了一种称为广义聚合易失性(Generalized Aggregation Proneness ,GAP)的算法。基于氨基酸残基对的倾向,GAP扫描给定氨基酸序列的淀粉样蛋白和无定形β-聚集六肽区段,以在参与聚集的β-链的相同或交替面处一起发生。使用现有实验数据进行的基准研究表明,GAP的表现水平远高于其他APR预测算法。例如,Tsolis等人最近编制了一组在33个促淀粉样蛋白中发现的48个淀粉样蛋白形成肽序列。这些序列用于对几种可自由使用的APR预测工具的性能进行基准测试。如果肽序列含有至少一个APR(六个或更多个连续残基被鉴定为聚集),则认为肽序列是淀粉样蛋白形成的。结果如表1.1所示。 GAP比其他方法准确得多。总之,上述讨论表明,可以从生物候选药物的氨基酸序列预测物理化学降解位点。可以使用生物候选物的结构模型进一步确定这种预测。
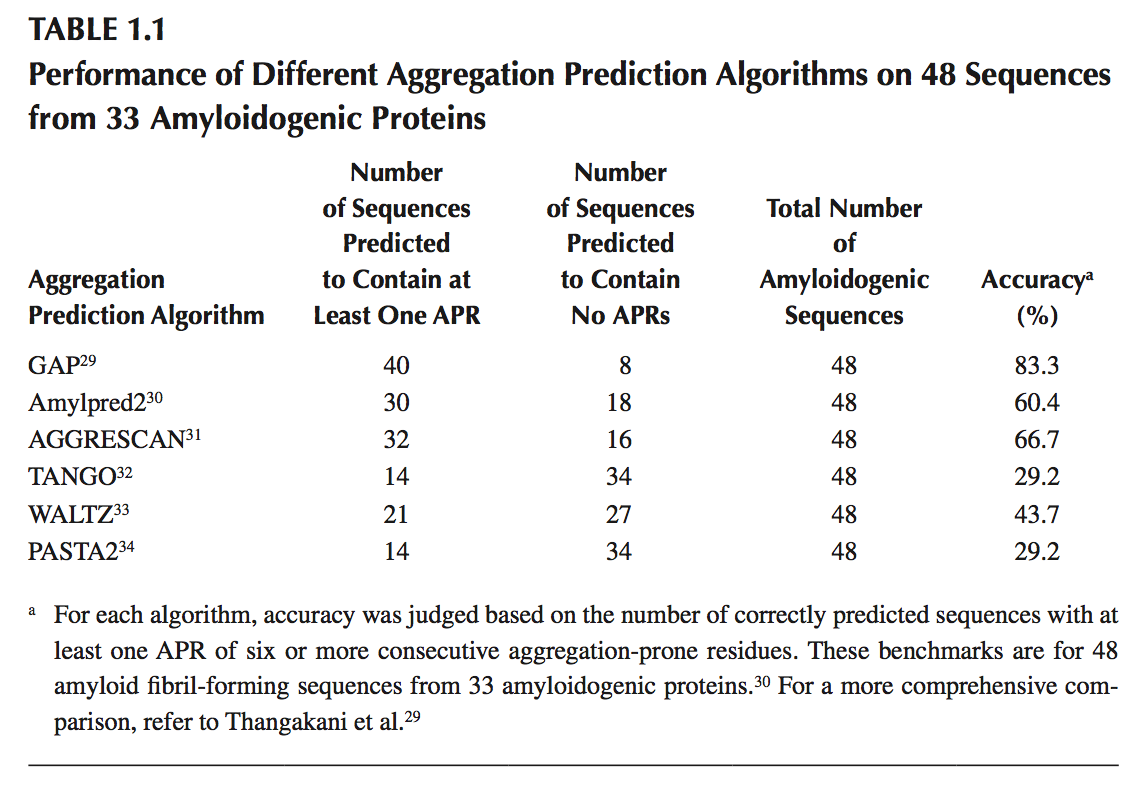
为了详细说明使用氨基酸序列和结构模型来预测生物候选物中潜在的物理化学降解位点(physicochemical degradation sites),使用人b12单克隆抗体,其全长晶体结构可在蛋白质数据库(PDB)条目1HZH中获得。 考虑到这是制剂开发初期的生物药物候选物,需要对潜在的物理化学降解途径进行评估。 图1.3显示了人b12mAb的氨基酸序列中潜在的物理化学降解位点,表1.2计算了mAb的可变区和恒定区中此类位点的数目。 图1.3和表1.2显示了人b12单克隆抗体中一对重链和轻链序列的潜在物理化学降解位点。 全长mAb包含两对这样的对。

图1.3 潜在的物理和化学liabilities映射在b12人mAb的序列上。理解图的关键如下:大胆的红色字体:潜在的化学降解位点(每种责任类型的详细计数见表1.1);下划线:自动预测的CDR。黄色背景:使用TANGO和PAGE的组合预测聚集倾向区域(Aggregation-prone regions ,APR);青色背景:WALTZ预测的其他APR。在一些情况下,TANGO / PAGE预测的APR与WALTZ预测的APR重叠。在这种情况下,在青色背景中仅显示另外的残基;灰色背景:通过使用基于淀粉样蛋白形成肽STVIIE的变体的实验衍生模式检测的另外的六肽聚集倾向区域。这些六肽模式中的一些与TANGO / PAGE和WALTZ预测的APR重叠。在这种情况下,仅灰色背景中显示额外的残基。绿色背景:聚糖附着位点。
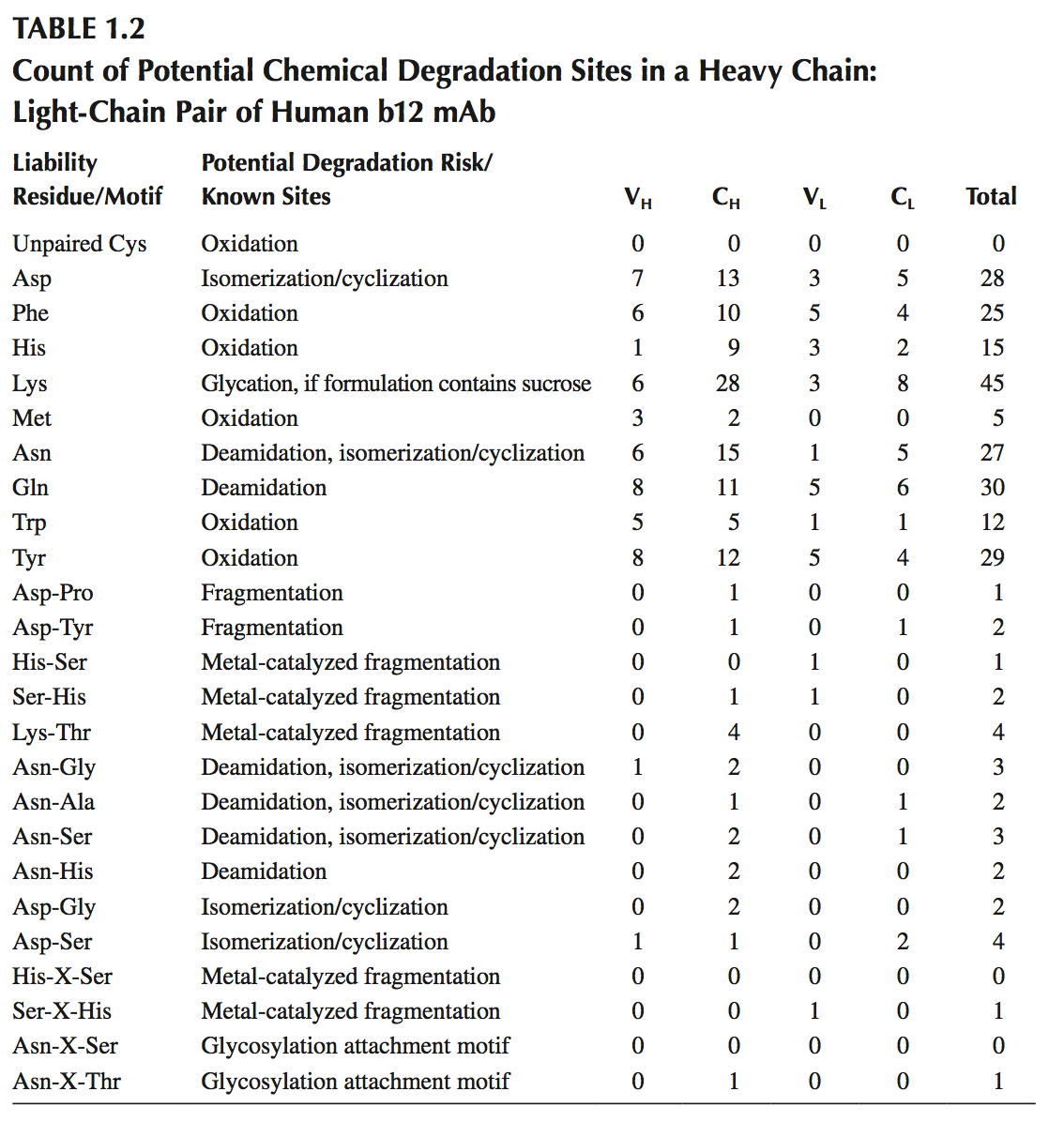
图1.3和表1.2通过指出大部分氨基酸序列固有地倾向于几种物理化学降解中的一种或另一种来说明大分子如mAb的复杂性。化学降解通常要求所涉及的残基存在于蛋白质表面上,使得它们可以与溶剂,金属离子,氧化还原剂等相互作用。类似地,APR需要位于蛋白质表面处或附近,以能够响应于诸如温度的物理应力而促进聚集。因此,建立三维结构模型对于确定哪些上述标记位点的物理化学降解风险更大是必不可少的。对于人b12抗体,全长抗体的晶体结构是公众可获得的。大多数生物制药候选药物通常无法获得3D结构。此外,结晶每个候选物及其变体是昂贵且耗时的。蛋白质结构预测的计算技术通常用于生物制药产品的研究和开发。如果有合适的模板,可以快速推导出基于同源性的生物候选模型。基于同源性的蛋白质结构预测是一个广泛的领域,在药物发现和设计方面有多种应用,该评论不在本章的范围内。简而言之,同源性建模依赖于具有相似氨基酸序列的蛋白质具有相似3D结构的前提。因此,它利用目标蛋白质的氨基酸序列与模板蛋白质的相似性,模板蛋白质的实验结构可用于模拟靶蛋白质的3D结构。近年来已经开发了抗体可变结构域(Fv和Fab)的计算建模程序,并且这些程序在基于抗体的治疗学的基于结构的设计中证明是有帮助的。基于同源性的抗体可变区和全长抗体模型可用于理解候选物的物理化学属性和精确定位潜在的物理化学降解位点。
让我们继续我们的人类b12抗体的例子和使用mAb序列进行的预测(图1.3)。 如前所述,该mAb的2.7Å分辨率晶体结构可在PDB条目1HZH中公开获得。 该结构具有一些缺失的残基和断裂的断裂的重链间二硫键。 这些是通过分子建模完成的。 全长mAb含有两对重链和轻链,其折叠成12个结构域,这些结构域又组织成两个Fab区和Fc区。 每个Fab含有重链 - 轻链对的变量(VL和VH组织成Fv区)和恒定(CH1和CL)结构域。 Fc区含有来自两条重链中的每一条的恒定结构域CH2和CH3。 Fc区中的CH2结构域也是糖基化的。 图1.4显示了人b12 mAb的3D结构的示意图。
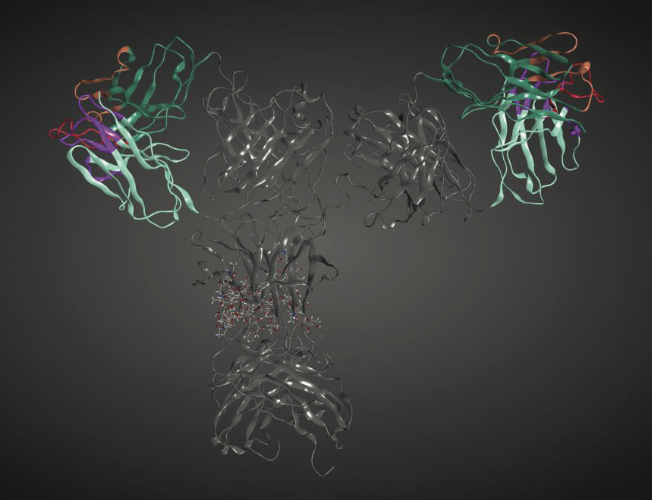
图1.4显示了全长人b12 mAb(PDB ID,1HZH)的示意图。 可变区域以不同颜色的条带显示。 在这些区域中,重链和轻链显示为绿色和青色色带。 重链CDR1和2以棕色显示,CDR3以红色显示。 轻链CDR1,2和3以品红色显示。 mAb中的恒定区以灰色条带显示。 聚糖以棒状显示。
让我们关注与该mAb中的互补决定区(CDR)相邻或重叠的物理化学降解位点。 涉及基于抗体的药物产品的这些区域的降解是直接关注的,因为它们可能潜在地损害产品的活性。 图1.3表明,β链介导的聚集,Asp异构化,Asn脱氨,Met氧化,金属催化断裂和糖化(如果配方缓冲液含有蔗糖)的潜在位点落在CDR内或与CDR重叠。 人b12 mAb结构模型的可用性有助于计算单个氨基酸残基的溶剂可及表面积和mAb在典型配方条件下的分子特性。 通过检查降解倾向位点的溶剂暴露,可以单独使用氨基酸序列进行预测:
-
Met氧化(oxidation)的风险:重链(H3环)中的CDR3含有Met 114(连续序列编号)。 H3环以主要方式促进抗原结合。 因此,序列信息表明,这些环中存在的Met残基的氧化可能会损害该mAb的活性。 然而,可及表面积(accessible surface area,ASA)计算显示Met 114在两条重链中都是溶剂不可接近的(Met 114的ASA = 0.0%)。 因此,Met氧化可能不是该mAb的风险因素,只要其3D结构得以保留即可
-
Asn脱酰胺(deamidation)的风险:该mAb中六个CDR环中的四个含有Asn残基。 特别地,重链(H2环)中的CDR2含有55-NG-56基序。 ASA计算表明该基序是溶剂暴露的,因此易于脱酰胺。 除此之外,H1环(N51)和L2环(N54)中的Asn残基也在重链和轻链的两个拷贝之一中溶剂暴露。 因此,如果Asn→Asp / IsoAsp取代干扰抗原结合,Asn脱酰胺可能是该mAb的风险因素。
-
片段化(fragmentation)的风险:轻链中的CDR1(L1环)含有金属催化的片段化基序26-SHS-28。 ASA计算表明该基序在两条轻链中都是溶剂暴露的。 因此,金属催化的片段化可能是该mAb的风险因素。
-
Asp异构化(isomerization)的风险:重链(H3环)中的CDR3含有Asp异构化基序,105-DS-106。 ASA计算表明该基序在两条重链中也是溶剂暴露的。 因此,如果Asp→IsoAsp代谢物干扰抗原结合,Asp异构化可能是该mAb的风险因素。
-
糖化(glycation)风险:将几种生物制剂中存在的蔗糖降解为还原糖,葡萄糖和果糖,可通过美拉德(Maillard)反应导致Lys残基糖化。该抗体重链(H2环)中的CDR2含有两个Lys残基 ,K58和K63。 ASA计算表明,这两种残留物均溶剂暴露,带电,并且pKa在Lys糖化的范围内。因此,如果制剂含有蔗糖和pH值,Lys残留的糖化可能是该mAb的危险因素,如此导致其倒置。
-
聚集(aggregation)风险:重链(H1,H2和H3)中的所有三个CDR和轻链(L3环)中的CDR3与APR重叠或包含APR。 ASA计算表明APR 28-RFSNFVIHWV-37中的一些残基与H1环重叠,是溶剂暴露的。 通过分子动力学模拟进一步探测了β介导的聚集问题。 这些将在下一节中介绍。
总体而言,上述练习演示了如何使用结构信息使我们能够排除几个基于序列的预测位点的物理化学降解风险,因为这些位点可能无法溶剂化。同时,这些信息还有助于我们突出可能特别危险的残留物和区域,因为它们位于生物候选物的三维结构中。此外,结构模型的可用性有助于识别单独的氨基酸序列可能不明显的潜在表面特征。例如,溶剂暴露的疏水补丁(patches)可以作为易聚集的motifs。 Chennamsetty等人开发并验证了一种称为空间聚集倾向(Spatial Aggregation Propensity,SAP)的算法来检测这种易聚集的基序(motifs)。商业的分子建模和模拟软件,例如来自Chemical Computing Group的MOE,也能够检测溶剂暴露的疏水和带电补片(patches)。例如,图1.5显示了溶剂暴露的带正电荷(蓝色),带负电荷(红色)和疏水(绿色)斑块,这些斑块映射到人类b12 mAb的分子表面。
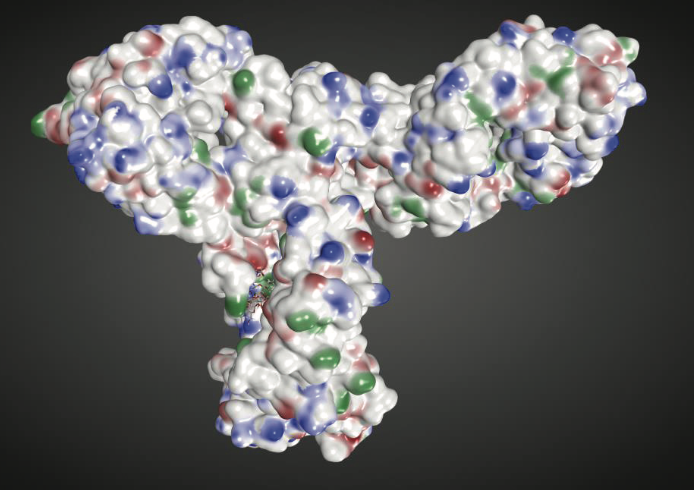
图1.5使用MOE中的Patch Analyzer检测人体b12 mAb上溶剂暴露的疏水性(绿色),带正电荷(蓝色)和负电荷(红色)斑块。
除了回答有关特定物理化学降解位点的问题之外,3D结构的可用性还可以计算生物制品候选物的几种有用的溶液特性,例如pI,电荷,体积,扩散系数和偶极矩。表1.3给出了b12 mAb及其组分的这些性质,即Fv(重链,Gln1-Ser127;轻链,Glu1-Lys108),Fab(重链,Gln1-Cys230;轻链,Glu1-Cys215),和Fc(两条重链,每条Glu243-Lys457和聚糖)区域。在pH 5.8和1mM盐浓度下计算这些性质。这些性质可用于估计生物候选物在稀释浓度下的溶液行为。例如,该mAb中的所有区域具有显着的正净电荷和ξ-电位值。它们的pI也高于配方pH。在这样的条件下,该分子的不同拷贝之间的胶体相互作用主要是排斥的,并且该mAb的高浓度制剂可能表现出低粘度。

在药物发现和配制的早期阶段获得这种详细的分子水平信息,可以以多种方式被使用。例如,可以在具有相似效力的几个候选者之间比较这样的信息,并且在主要候选人选择期间考虑这些信息。该信息还有助于从具有相对较差的物理化学稳定性中,重新设计出的高效先到候选物。特别是,消除潜在的聚集位点可以提高生物候选物的溶解度。在分子序列是固定且不能改变的配制阶段,该信息可用于制定配方策略。例如,如果CDR中存在溶剂暴露的Met残基,则在配制缓冲液中添加甲硫氨酸作为抗氧化剂以减轻氧化产生的效力损失可能是有用的。类似地,如果存在可能影响效力的潜在金属催化的氧化和断裂位点,则添加螯合剂可能有所帮助。最重要的是,事先获得这些信息可以节省配方开发研究所需的时间和材料。
四、利用计算推进我们对生物制药物理化学降解的认识
生物制药候选物通常表现出与低热力学稳定性,溶解性差,聚集倾向和浓缩溶液中的高粘度(high viscosity)相关的问题。 在生物制剂所显示的所有物理化学降解中,聚集是最常见的,在纯化,配制,运输和储存过程中会遇到多次(图1.2)。 除了在纯化步骤中材料的损失之外,聚集还可能降低效力并增强与生物药物相关的免疫原性风险。 与折叠类似,蛋白质聚集已成为生物化学中一个主要未解决的问题。 它在蛋白质操作过程中经常在实验室中遇到,并且它涉及几种人类神经退行性疾病。 有关生物制剂聚集的综合概述可在其他地方获得。在这里,我们总结了我们使用数据分析和分子模拟来理解单克隆抗体聚集的进展。
蛋白质分子可以以几种方式彼此结合,包括由于表面暴露的疏水性热点的排除溶剂(水)的倾向,形状以及相互作用的蛋白质表面之间的电荷互补性,以及β-链介导的聚集体。其中,β-链介导的蛋白质聚集可能需要使天然蛋白质结构不稳定,并导致不可逆的无定形β-聚集体或类淀粉样蛋白,具有良好的形态特征。 β链介导的蛋白质聚集已被证明是人类中几种神经退行性疾病的基础,涉及形成交叉β空间拉链基序( cross-β steric zipper motif )容易通过荧光染料染料刚果红和硫代蛋白T(ThT)检测。在一项开创性的研究中,Maas等表明生物药物经过有效期也可以形成β-链介导的聚集体,结合ThT,并引发免疫反应。该观察结果促使Wang和同事将从致病淀粉样蛋白的疾病研究开发的APR预测程序,应用于市售治疗性抗体的序列。在商业上可获得的治疗性mAb序列中发现了能够启动β-链介导的聚集的几种重复的APR。此后已经通过实验证明,当暴露于诸如酸变性或升高的温度的应激条件时,mAb确实形成可使用ThT结合测定检测的β-链介导的聚集体。
Wang等人的研究中有趣的观察结果是,APR经常与治疗性mAb的CDR重叠。该观察结果提出了APR中发现的残基也可能参与抗原结合的可能性。 通过对在PDB中可获得高分辨率晶体结构的29种非冗余Fab-抗原复合物的分析,发现APR大约贡献了与抗原接触的Fab表面的1/5。 聚集和抗原识别之间这种偶联背后的原因是在APR和CDR中都发现芳香族残基Tyr和Trp的倾向。 这项工作提出了几种合理的基于结构的设计或选择策略,以提高基于抗体的生物制剂的可开发性。 这些合理策略的目标是通过破坏战略性选择的APR,来减少聚集并改善基于抗体的治疗剂的溶解度和高浓度溶液行为。
必须了解APR对蛋白质天然状态的贡献,以便选择可能适合于破坏的APR。除促进聚集外,APR还在蛋白质结构功能中发挥其他几种作用。最近使用包含随机产生的氨基酸序列,蛋白质家族,固有无序蛋白质,蛋白质单体和催化残基的多个数据集研究了这些作用。发现单体蛋白质中的氨基酸序列比使用相同氨基酸组成产生的随机序列具有更低的聚集倾向,这意味着天然蛋白质序列中的聚集的进化偏差。 APR对蛋白质稳定性的贡献大于相似大小的片段的平均值,并且在催化残基的结构附近比偶然机会更常见。这些观察结果表明,APR促进结构有序,因此在本质上无序的区域中发现较少。有趣的是,蛋白质已经进化,以优化其在细胞中聚集的风险。优化策略包括从同源蛋白质序列家族收集的两种相反趋势。一方面,寻求在密切相关的蛋白质序列中最小化APR的发生率,另一方面,对于蛋白质折叠和功能重要的APR在更多不同序列中是保守的。这些研究的结果导致了通过破坏APR改善蛋白质溶解度的合理策略
选择用于破坏的APR的另一个方面是知道APR是否位于蛋白质表面处或附近,并且在蛋白质结构响应于物理应力而不稳定的情况下将主动促进聚集。这些研究通常涉及蛋白质结构的动态方面,而分子动力学(molecular dynamics,MD)模拟是检验这些的有用工具。通过在高温下对生物候选物进行MD模拟,我们可以尝试模拟在蛋白质制剂开发研究期间进行的加速稳定性实验。总之,还可以使用MD模拟研究抗体构象动力学以及聚糖截短和去糖基化对基于抗体的治疗剂的稳定性的影响。图1.6显示了全长鼠IgG2a抗体的完全溶剂化的分子系统(大约400,000个原子),其晶体结构可从PDB条目1IGT获得。下面,我们总结了从抗体的分子模拟中获得的见解
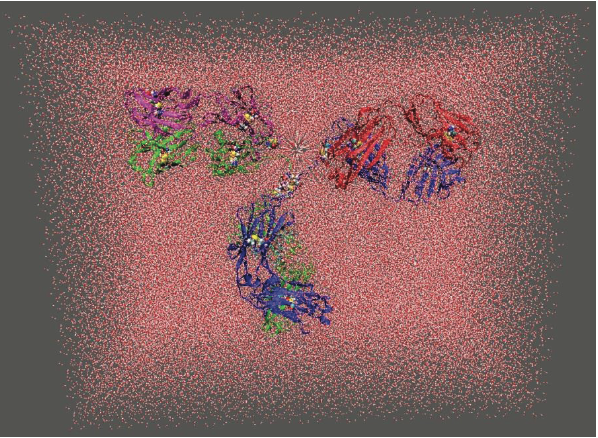
图1.6由Wang等人模拟的完全溶剂化的IgG2a鼠抗体分子系统。除了用CPK表示的二糖桥和聚糖外,mAb以带状显示。
分子动力学(Molecular dynamics ,MD)模拟用于研究全长鼠抗体(1IGT)在去糖基化(de-glycosylation)和热应激(thermal stress)时的构象去稳定化。Cα-原子均方根偏差(root mean square deviation ,RMSD)和骨干均方根角(root mean square uctuation,RMSF)计算用于研究在模拟过程中mAb结构的变化。发现由于去除聚糖残基导致的mAb去稳定化是局部的。它仅限于CH2结构域的四级和三级结构中的扰动。另一方面,热应力引起Fab和Fc区的广泛不稳定。我们的目的是使抗体不稳定,但不会展开。因此,在模拟期间没有观察到大规模的结构熔化。有趣的是,热应激和去糖基化似乎可以识别当抗体的天然结构不稳定时可能变得活跃的不同APR。预测存在于CH2结构域的链A中的APR在去糖基化后变为溶剂暴露。在热应激的情况下,存在于两个CDR重叠APR中的残基显示出增加的RMSF和平均溶剂暴露的变化。
全长鼠mAb的MD模拟之后是对人b12 mAb的Fab部分的类似模拟,其基于序列和结构的潜在物理化学降解位点已在上文中描述(参见第2节)。 Fab是比全长mAb小得多的分子系统。这使我们能够更详细地研究Fab动力学行为。与APR一起,研究了各个结构域及其界面的变形以响应热应力。变量(VH:VL)和恒定域(CH1:CL)界面在各个结构域的熔化之前变形。这在不同的模拟轨迹中始终如一。有趣的是,存在于结构域界面的β-链比Fab中的所有其他β-链和环更耐受熔化更长的持续时间。因此,存在于域接口区域中的APR不太可能参与该mAb的聚集。该研究表明CDR,与CDR相邻的构架区β-链和边缘β-链基本上在热应力下变形,导致这些区域中存在的APR的溶剂暴露。进一步分析允许基于溶剂暴露,结构位置,移动性和守门残基的发生率,排序不同序列预测的APR以促进其不可逆的β-链介导的聚集的能力。发现两种预测的APR,28-RFSNFVIHWV-37(与重链中的CDR1重叠)和122-TTVIVS-127(从VH转变为CH1结构域的序列区域),被发现特别危险。综合第3节关于b12抗体的序列和结构以及上述MD模拟的观察结果,可以得出结论,使用结构和动力学信息可以帮助重新预测生物候选药物中的物理化学降解位点。 从上述研究中获得的见解,在设计基于结构的分子设计策略或选择具有改善的可开发性属性的基于抗体的生物制剂方面非常有用。
五、生物学候选物不良免疫原性的计算评估
接受生物药物治疗的患者产生的不良免疫反应会导致生物药物的安全性和有效性担忧。在过去的三十年中,免疫安全问题已经指导治疗性mAb产品从小鼠来源到嵌合体到完全人源化。然而,含有完全人类抗体或重组/血浆衍生的人类蛋白质的生物制药产品仍然可以引起患者的不良免疫反应。
由于在致病微生物和共生微生物存在下维持体内平衡的需要而产生的免疫系统的复杂性质,不可避免地导致许多潜在的促成蛋白质免疫原性的促成因素。虽然对产品的免疫原性风险的总体评估必须考虑几个个体风险因素,但通常认为T细胞表位(T-cell epitopes)代表了对这些不需要的免疫反应的显着贡献,因为它们是类别转换(class switching)和B细胞显着扩展(signi cant expansion )所必需的。此外,由于T细胞反应在免疫中的核心作用,已经进行了多次尝试以通过能够鉴定可能与主要组织相容性复合物(MHC)II类(人白细胞抗原[HLA])结合的肽的算法,预测蛋白质氨基酸序列中存在的T细胞表位。这些算法最初应用于微生物蛋白质作为疫苗设计策略的一部分,但现在经常用于分析生物候选物的氨基酸序列。
当应用这些工具来评估免疫原性风险时,必须注意确保序列是外来的(例如,工程化mAb的CDR或接受者中缺失的内源蛋白质)或者反应性显着可能性将存在T细胞。这可以通过例如健康供体或患者样品中的有害T细胞测定来完成。应该用mAb进一步试验以确定种系序列是否真正存在于群体中,因为患者很可能接受治疗将具有不同的种系序列,因此对于构成异物的内容具有不同的比例。这里,将这些考虑因素应用于人b12抗体(PDB entry,1HZH)的氨基酸序列作为实例(图1.7)。可以看出,潜在的HLA结合肽存在于整个抗体重链和轻链中。然而,基于外来概念的来自这些序列的潜在T细胞表位仅为三个。通过体外试验确定预测很常见。这些通常是HLA结合测定,但是涉及测量由DC处理和呈递的肽的新一代测定正在出现。

图1.7人b12抗体(PDB entry,1HZH)的氨基酸序列中预测的T细胞表位。 此示例表明Epivax EpiMatrix算法进行的预测。以黄色突出显示的序列区域是潜在的HLA结合肽,以红色突出显示的序列是含有外源(非种系)残基的潜在HLA结合肽,并突出显示序列 绿色是生殖系起源(germ line origin)的潜在HLA结合肽,并且被认为可能上调保护性T调节细胞。 VH和VL区内的粗体残基与亲本种系序列(parental germ line )(VH,IGHV1-3 * 01; VL,IGKV3-20 * 01)不同。
免疫原性的另一个潜在风险是B细胞表位(B-cell epitopes)的存在。用于鉴定B细胞表位的预测算法落后于T细胞表位的预测算法,可能最显着的是由于T细胞表位是线性的。相反,许多B细胞表位仅出现在蛋白质的三维结构中(构象表位)。然而,确实存在评估线性和构象B细胞表位的一些努力,尽管当针对已知表位判断时它们的预测能力范围在30%和50%之间。至于这些方法的验证,实际上对于治疗性蛋白质来说很少有B细胞表位,这使得该空间中任何算法的训练集相当有限。一个例子是关于阿达木单抗(adalimumab)的数据,表明B细胞表位存在于CDR中。尽管尚未确定确切的表位,但几乎完全针对CDR的抗药物抗体(anti-drug anti-body, ADA)反应性的检测应该是关于如何在将来开发这些算法的有用指南。图1.8显示了人b12抗体中潜在的线性B细胞表位。注意,预测外源性线性B细胞免疫表皮位于重链的CDR2和人b12抗体轻链的框架区2中。设计用于预测不连续B细胞表位的算法,Discotope,也在与外源线性B细胞表位相同的区域中鉴定潜在的B细胞表位。这些discotope如图1.9所示。通过两种不同算法在人b12 mAb的相同序列-结构区域(重链中的CDR2,轻链中的FR2)中对B细胞表位的一致预测给出了更大的信心,即这些区域可能参与不良免疫反应如果这种抗体是作为药物产品开发的话。然而,需要进一步表征抗药物抗体(ADAs)以确定当前B细胞表位预测的准确性。这将反过来导致预测算法的改进。

图1.8 在人b12抗体(PDB entry,1HZH)的序列中通过Emini表面可及性算法预测线性B细胞表位。 以黄色突出的序列区域是自身表位,而红色的序列区域是外来B细胞表位。 VH和VL区内的粗体残基与亲本种系序列(parental germ line sequences)(VH,IGHV1-3 * 01; VL,IGKV3-20 * 01)不同。
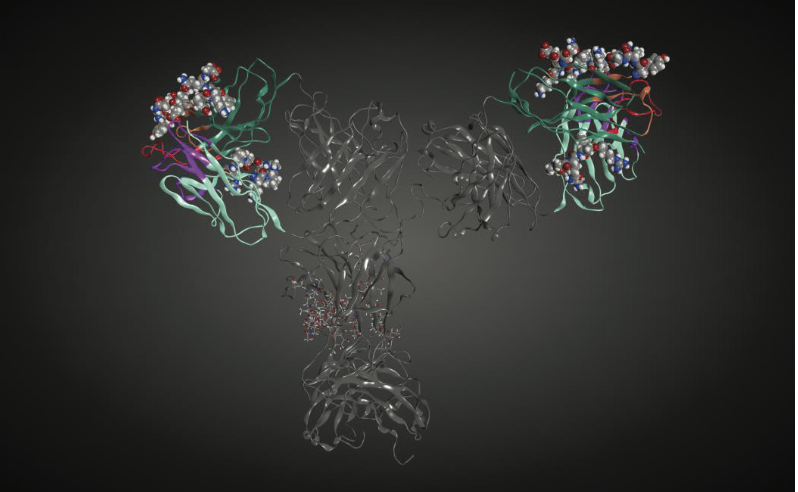
图1.9 在人b12抗体(PDB entry,1HZH)的3D结构中通过Discotope预测B细胞表位。 参与蛋白质表位的残基用CPK表示。 这里仅显示与外源线性B细胞表位一致的那些同位素。
将免疫原性风险评估与免疫生物学原理相结合的多方面算法可以促进人免疫系统的预测系统生物学模型的生成。 存在几种这样的模型(例如,参见Lee等人),尽管很少有模型应用于免疫原性和预测结果的问题。 最近尝试用adalimumab做这件事就是可能的。 我们目前是否对免疫系统中的关键参与者产生真正的预测模型有足够的了解还有待观察,尽管这些模型可能会提供一个框架来产生假设和指导免疫原性评估。
六、理化学降解与生物安全的联系:聚集 - 免疫原性耦合 (LINKING PHYSICOCHEMICAL DEGRADATION WITH SAFETY OF BIOLOGICS: AGGREGATION– IMMUNOGENICITY COUPLING)
生物制药以高选择性结合其靶标。这是一个主要优点,因为在这种情况下非机械毒性(副作用)大大减少。然而,除了上面讨论的短序列区域的外来性之外,生物制剂的物理化学降解的免疫学后果还导致生物制剂的安全性和有效性问题。还必须强调的是,产品质量只是导致接受这些疗法的患者中不希望的免疫反应的众多因素之一。其他因素包括患者的遗传构成和医疗历史。已经证明几种生物药物产品的施用会导致免疫后果,其中许多可能与产品质量属性有关,例如聚集。免疫学后果的临床表现是产生针对生物制剂的抗药物抗体(ADAs)。 ADA可以定位生物制剂并降低其效果,甚至可以使它们随着时间的推移无效。生物制药产品中存在的聚集物如何引发免疫反应尚不完全清楚。但是,似乎涉及T细胞介导的适应性免疫。 如上所示,使用它们的氨基酸序列预测生物药物候选物中潜在的β介导的聚集倾向区域(aggregation-prone regions,APR)以及T细胞免疫表位(T-cell immune epitopes,TcIE)是可行的。与APR一样,TcIE也经常在治疗性抗体的CDR和邻接区域中发现。那么问题就是,APR和TcIEs蛋白质序列是否重叠?如果他们这样重叠了,这对生物制药产品的质量和安全意味着什么?最近,我们研究了市售治疗性mAb中APR与HLA-DR结合TcIEs7之间的潜在偶联。发现大多数APR落在重叠的TcIE区域(TcIERs)内,其能够混杂地结合几种MHC II类等位基因。这些观察结果表明,共同的序列结构基序有助于这些生物制剂的物理化学降解和免疫原性。因此,同时减轻生物候选药物的聚集和免疫原性风险是可行的。同时优化药品质量和安全属性将为下一代生物药品铺平道路。
已知参与自身免疫和神经退行性(neurodegenerative)疾病的几种人类蛋白质也显示出聚集和免疫原性之间的耦合。 APR和TcIE之间重叠的这种证据可在人胰岛素的晶体学水平上获得。 来自胰岛素B链的肽已经与MHC II类等位基因HLA-DQ(PDB entry,1JK8)复合结晶。 共晶中的肽含有淀粉样蛋白形成部分11-LVEALYL-17(PDB entry,3HYD75)。 图1.10显示MHC-胰岛素B链复合物,其中突出显示淀粉样蛋白成束(amyloid bril-forming portion)部分。 一直以来,已经观察到,连续皮下输注胰岛素治疗的患者可能由于针对泵系统中存在的胰岛素提取的抗胰岛素IgG抗体而产生过敏。 一种称为注射部位淀粉样变性(injection site amyloidosis)的罕见人类疾病也是由频繁注射部位的胰岛素形成引起的。

图1.10 显示了聚集和免疫原性偶联的实例。 显示了与胰岛素B链复合的MHC II类等位基因HLA-DQ8的结构(PDB entry,1JK8)。 肽中的所有残基和HLA-DQ8中的附近残基以球和棒显示。 胰岛素B链中的黄色部分也显示出形成淀粉样蛋白凝胶(PDB entry,3HYD)。
金属离子结合位点也可位于生物制剂中的APR附近。 最近观察到,增加mAb溶液中Fe3 +离子的摩尔浓度会导致mAb中的氧化和聚集水平增加。 对mAb结构的检查揭示了整个结构中的几个Fe3 +结合位点及其簇,包括CDR和铰链区。 特别地,在mAb的CDR中观察到Fe3+结合残基(Asp和Glu),芳香族残基(Trp,Tyr和Phe)和APR的共定位(图1.11)。 金属催化的芳族残基的氧化可能使该mAb不稳定,导致聚集和潜在的免疫原性。
七、通过计算估算生物候选物的高浓度溶液行为 (ESTIMATING HIGH-CONCENTRATION-SOLUTION BEHAVIOR OF BIOLOGICAL CANDIDATES VIA COMPUTATION)
从患者的方便性(convenience)和规范性(compliance)的角度来看,能够提供小体积(~1ml)的高浓度(~100-150 mg/ml)生物药物溶液最好不过了。然而,高粘度(viscosity)可能是成功开发高浓度(high-concentration )生物制药产品的重要障碍。
需要快速识别生物药候选物,但识别高浓度候选物比较困难。用于鉴定此类候选物的计算工具在药物发现和制剂的早期阶段是有价值的。要开发此类工具,不同mAb上可用的实验数据必须一致。也就是说,生物分子在相同的条件下使用尽可能相同的实验程序进行研究。例如,Li等人最近获得了在不含添加盐的相同缓冲液中配制的11种治疗性mAb候选物的粘度数据。所有粘度测量均使用Anton-Paar粘度计使用相同的程序进行。使用相同的计算方法从氨基酸序列和这些mAb的Fv部分的结构模型计算几种分子性质,例如净电荷,pI,ξ-电位,疏水性和聚集倾向。这促进了统计相关性分析和分子谱的建立。该分析证实了这样的假设:可变区的分子特征在确定mAb的高浓度溶液行为中起重要作用。
这是由于以下原因:mAb的高浓度溶液行为必须来自胶体分子间相互作用的性质和它形成的自缔合。分子间相互作用由mAb的氨基酸序列和3D结构特征决定。对于所有mAb候选物,仅涉及人mAb的恒定区的胶体(colloidal)分子间相互作用可能是相同的。因此,从mAb变为mAb的可变区(Fvs)也调节它们的胶体分子间相互作用。在上述研究中显示150mg/ml高粘度的mAb的Fv部分在配制缓冲液pH值5.8下具有显着的负净电荷和负ξ-电位。
Fv区的pI值也低于制剂缓冲液pH。这些观察结果有助于基于可变区($Z_{Fv}$,$ξ_{Fv}$和$pI_{Fv}$)的净电荷,ξ-电位和pI的计算值来设计mAb候选筛选方案。这种方法可用于“red fagging”治疗mAb候选物,其作为高浓度药物产品的发展可能受到与粘度相关的挑战的阻碍。图1.12显示了通过在不同mAb中呈现Fv区域的静电表面的这些特征的实例

图1.11 显示了mAb的Fv部分的CDR中Fe3 +离子结合残基,芳族残基和APR的共定位。 重链中溶剂可接近的Fe3 +离子结合残基E46,D54和D62以球 - 棒表示,以及它们的近端芳族残基W47,W52,W53,Y60和Y106描绘。 注意残基根据它们在序列中的位置编号。 使用以下颜色代码:重链,绿色; 轻链,红色; 和APR,黄色。

图1.12显示了Fv区域的表面静电图示例,这些图谱属于从11 mAbs44的浓度依赖性粘度行为分析推导出的三个剖面中的一个。顶行显示了跟随分布1的Fv区域的示例(显着正净电荷和Fv区域的ξ-电位)。即使在高浓度下,这种mAb通常也具有低粘度。中间一行显示Fv区域的静电表面遵循轮廓2(Fv区域上的净电荷几乎为零)。在这种情况下,疏水性分子间相互作用发挥更重要的作用,并且目前难以可靠地预测这种mAb的浓度依赖性粘度行为。来自谱3的mAb的Fv区域(在制剂pH下具有显着的负净电荷和ξ-电势以及在制剂缓冲液pH以下的pIFv)显示在底行。该分布中的mAb通常在高浓度下显示高粘度,因为静电和疏水分子间相互作用促进可逆的自缔合。在所有这些表面静电图中,越来越多的蓝色和红色分别表示越来越多的正电荷和负电荷。
生物候选物的溶液行为的多尺度模拟可以产生对它们的胶体(colloidal)分子间相互作用的见解。这种模拟需要由数百至数千个溶质拷贝(生物制剂)组成的分子系统。为了加速这种模拟,生物制剂的分子结构特征是粗粒度(coarse-grained,CG),并且仅粗略地考虑溶剂。 Chaudhri等开创了高度集中的抗体解决方案的CG模拟,为他们的自我关联行为提供了宝贵的见解。图1.13显示了人IgG1b12抗体的CG模型。在该模型中,抗体的每个结构域通过球形CG interaction bead近似。对于b12抗体的每个结构域,将单CG interaction bead分配给质心。每个珠子(bead)的质量和电荷分别是珠子所分配的区域的质量和净电荷。总之,每个mAb分子具有12个CG相互作用珠。因为可变结构域在每个mAb中是不同的,代表这些结构域的四个CG相互作用珠子从mAb到mAb不同。

图1.13 显示了人IgG1 b12 mAb的粗粒度(coarse-grained)表示。这种粗粒化有利于抗体溶液行为的多尺度模拟。

图1.14 A typical coarse-grained simulation box is shown. Each antibody molecule is represented by a 12-bead coarse-grained model.
图1.14显示了一个典型的CG模拟框。 CG模拟用于比较来自上述11种mAb数据集的三种mAb(mAb 1,mAb 10和mAb 11)的浓度为30和100mg / ml的扩散行为。在这三种中,mAb 1表现良好(浓度高于100 mg / ml时粘度低),而mAb 10和11表现不佳(浓度高于100 mg / ml时粘度高)。使用LAMMPS85进行模拟,并在具有周期性边界条件的真空中在300K下运行5μs,时间步长为1ps(10-12s)。这些模拟显示,在两种浓度下,mAb 10和11的扩散明显慢于mAb 1的扩散。由于针对mAb 10和11的负电荷越来越多的Fv区域,Fab和Fc片段之间的静电互补性增加。这导致这些mAb的自缔合行为增加。还发现较高浓度的分子拥挤显着影响三种mAb的扩散。
八、结论和未来方向 (CONCLUSIONS AND FUTURE DIRECTIONS)
生物制药信息学(Biopharmaceutical informatics)是一个新兴领域,旨在推动各种计算工具的应用,如信息学,数据分析,预测建模和数学建模,以发现和开发生物制药药物。 在这里,我们举例说明了生物制药信息学如何帮助我们理解基础序列结构基序,这些基序驱动物理化学降解,免疫原性或生物制药的高浓度溶液行为。 该研究有助于为生物制品开发合理的设计策略,提高分子完整性和安全性。可能只需要蛋白质序列中的微小调整,不会影响效力。 这种设计策略非常需要提高生物制剂的成本效益并防止晚期药物失效。
预计生物制药信息学将在可预见的未来在生物药物发现和开发过程中得到越来越多的应用。这些应用将使生物药物发现和开发周期更加有效和灵活,从而使产品具有更高的稳定性,安全性和易用性。预计化学,制造和控制(CMC)方面的改进导致发现转化为市场上可获得的生物药物产品的成功概率更大,从而大大降低药物开发成本。这反过来将导致更多地利用生物制药来治疗人类疾病和增加市场份额。我们增加的生物药物集体经验要求建立公共和专有数据库,其中包含有关细胞系表达,生物物理稳定性,免疫原性,PK / PD和药物安全性的自洽实验数据,以及生物学的计算分子特性分子。此类数据库应能够挖掘出理想的药品特性,并提出将其纳入未来生物药物候选药物的策略。需要开发用于在体外和体内模拟生物药物分子的溶液行为的可靠计算方法。鉴于大量生物候选药物分子以及我们对蛋白质折叠,蛋白质 - 蛋白质相互作用和聚集的基本生物化学的不充分理解,实验和计算生物物理科学的进展有很多机会。这种研究需要优先考虑,因为它具有很大的实用价值。例如,在药物发现项目的早期阶段筛选抗体库中的氨基酸序列和优选结构模型以及潜在物理化学降解位点的下一代测序(NGS)输出,可以显着降低生物药物发现中的磨损风险和发展周期。在早期配方稳定性研究中,快速可靠地模拟生物候选物在各种配方缓冲液,pH值和赋形剂中的构象稳定性的能力可以显着降低对pH,缓冲液和赋形剂筛选实验的需求。
总结
本章描述了计算工具和方法在生物制药产品开发中的各种应用。引入了一个新术语 - 生物制药信息学(biopharmaceuti- cal informatics)。生物制药信息学是一个新兴的领域,科学家们试图了解生物制药产品开发过程中面临的挑战, 通过计算工具和方法分析序列和结构的特点,从而了解生物药物产品(如重组人类蛋白质,单克隆抗体和抗体药物偶联物等。在生物药物发现和制剂的早期阶段,计算建模和模拟的应用可以促进生物制药候选物的可开发性风险评估。当有多个候选物可用时,这种基于风险的评估可用于确定候选物的发展优先次序。或者,这些评估可用于优化生物制药候选物,以实现具有成本效益的药物开发,并通过分子重新设计提高安全性。在多个药物开发项目中收集的实验数据也可以分析,以及与候选物的分子属性的相关性,从而更好地了解理想的蛋白质序列结构特征,这可能使生物药物开发有效且具有成本效益。随着这些工具的成熟及其应用在生物技术和制药行业中的应用越来越广泛,预计生物制药药物开发的过程将变得更加有效。虽然本章重点介绍最成功的一类生物制药,即单克隆抗体,但这里介绍的工具和方法也可以应用于其他方式。
参考资料
- 《Developability of Biotherapeutics》
