【5.2】CDS和ORF,EXON,转录本,cDNA
简单总结:
Exons = gene - introns
CDS = gene - introns - UTRs
CDS = Exons - UTRs
蛋白质编码基因至少包含四个组件:TU, MODEL, EXON, CDS
- TU代表转录单位,是基因的最高顺式元件。仅在可变剪接体存在时可编码多个基因MODEL
- 基因MODEL封装了单个剪接异构体所有的的编码和非编码结构,每个基因MODEL可编码剪接数个mRNA EXONS并代表基因剪接后无内含子的部分。
- 一个mRNA EXON可能仅部分编码蛋白质,恰恰是上游或下游非编码区存在时。每个EXON的蛋白质编码部分由CDS元件表示。CDS元件还编码终止密码子。基因组件不是根据它们的坐标进行排序的。
- 对于存在非翻译区的区域,将出现UTR。UTR代表了RNA EXON的非蛋白质编码部分。目前,UTRs不支持该DTD以外的TIGR数据类型,它们的存在仅为了便于外部数据分析。
一、CDS与ORF的区别?为什么CDS可以包括很多个开放阅读框?
- 基因的编码区(Coding region),亦称为“编码序列”(Coding sequence)或“CDS”(Coding DNA Sequence),是指mRNA序列中编码蛋白质的那部分序列。
- CDS也等同于ORF(open reading frame)是编码蛋白质的序列,以ATG开始–终止密码子结束。
CDS的定义是对的,即CDS是已知的一个基因上确确实实翻译成蛋白质的区段。 ORF则是指,任意一段序列,只要起于ATG止于终止子,都可以叫做ORF。ORF是一种预测,而不是一种已知的翻译区。即随意写下一段DNA序列,只要以三个碱基为单位能找到ATG和终止子,就可以称作ORF,这段ORF甚至可能不是一段真正存在的DNA序列,但是它仍然是ORF。一段序列是可以有多个ORF的,只有当ORF符合已知的可翻译成蛋白的序列时,才能等同于CDS。

图中序列为 ATGCAATGGGGAAATGTTACCAGGTCCGAACTTATTGAGGTAAGACAGATTTAA 假设这是某种基因的CDS。那么在这段序列中,由图所示,会出现三种始于ATG终于终止子的片段,由于这三种都有可能翻译成氨基酸,所以你可以说这段序列有三种可能的ORF。但是在基因中真正翻译出来的只有1,那么当你说这段基因的CDS的时候,只能是1.
- 基因经过转录形成Pre mRNA,这里面包含着内含子和外显子(5端是以外显子打头,但是这段外显子不仅包含CDS,还包含5' UTR;3端是以外显子结束,但是这段外显子不仅包含CDS,还包含3' UTR),经过剪接形成成熟mRNA,内含子已减掉,如果抛开后来加上去的cap和poly A的话,这时全是外显子,但是不全是CDS,因为只有中间的那部分以起始密码子开始、以终止密码子结束的片段才是CDS,只有这部分才会被翻译成蛋白质。
- CDS是指被翻译成蛋白质的片段(故而肯定是以起始密码子开始,以终止密码子结束的片段),而ORF仅仅是指以起始密码子开始,以终止密码子结束的片段,可以说是潜在的CDS
,
ORF-----translation,
CDS----transcription
translation 是理论上的,而transcription则显然是事实存在的。
二、转录本
- 5’UTR :region at the 5' end of a mature transcript (preceding the initiation codon) that is not translated into a protein
- 3’UTR: 3‘端非翻译区,也就是mRNA两端的非编码片段
我们平常通过数据库查找某个基因的相关信息时,会发现该基因有多个转录本。为什么一个基因可以有多个转录本呢?转录本能干什么?
转录本其实就是基因通过转录形成的一种或多种可供编码蛋白质的成熟的mRNA。
一个基因有可能有多个转录本,原因是由于不同的剪接方式造成的。我们都知道,基因转录之后,首先是形成前体mRNA,通过剪切内含子连接外显子,5’端加帽及3’端加尾之后形成成熟的mRNA。
但是在剪切的过程中可能会剪切掉外显子,也有可能保留部分内含子,这样就形成了多种mRNA即多个转录本。
举个栗子:这是一个三个外显子两个内含子的基因结构图
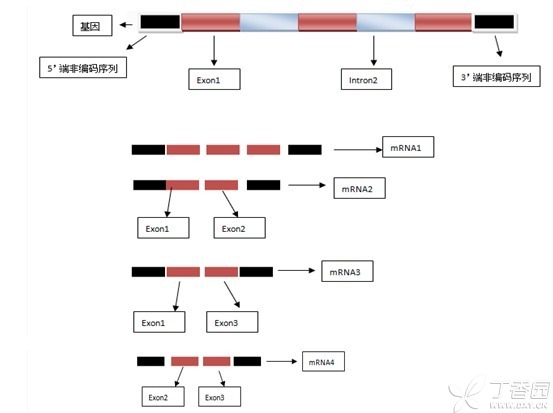
该图通过不同的剪接方式得到了四种mRNA即四种转录本(我只是列出了部分可能性),实际中可能该基因只具有其中的一种或两种转录本,也有可能都具有。
我们需要特别注意的是大多数基因有多个转录本,而且有可能每个转录本都会编码产生相应的蛋白,这样就有可能造成一个基因有多种功能。
我们平常研究某个基因时(该基因有多个转录本),其实我们研究的是它的其中一个转录本所编码的蛋白的功能。虽然该基因有多个转录本,而且每个转录本都编码蛋白,但是一般情况下它的不同的转录本分布在不同类型的细胞中,当然也有可能多种转录本同时存在于某一细胞中。
那我们研究该基因时应该怎么做呢?
首先,我们需要确定我们应该研究该基因的哪个转录本。
因为我们平常研究某个基因的功能的时候,是因为该基因在某一特定的组织和细胞中表达,它在这些组织和细胞中具有特定的功能,所以我们只需要确定该基因的哪个转录本在这些组织和细胞中表达即可。
确定的方法当然就是设计每种转录本特异性引物,然后通过RT-PCR就可知道哪种转录本在组织和细胞中特异性表达。那这个转录本就是我们接下来要研究的。
之所以要确定我们应该研究哪个转录本,那是因为它关系到引物的设计以及蛋白分子量的计算。
当我们研究某个基因的功能时,通常会抽提总的RNA,然后反转录得到cDNA,然后将cDNA连接到表达载体中转化到原核或真核细胞中进行表达,然后进行接下来的研究。
通过反转录获得cDNA时,引物的设计就是根据转录本设计的。而且之后我们会将表达的蛋白跑电泳后进行分析,那蛋白的大小是如何计算的呢,当然也是通过该转录本编码的蛋白的氨基酸序列计算的啊。
至于转录本的查询,也非常简单。在pubmed gene一栏输入目的基因,出现的页面下拉,发现如下
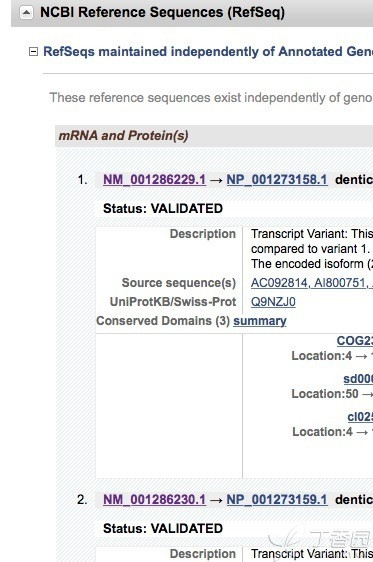
有多少个NM-号即有多少个转录本。先确定转录本才能进行引物设计
其他
cDNA:
cDNA是以mRNA为模板,在适当引物的存在下,由mRNA经过反转录而得到的DNA,是mRNA链互补的DNA链,其内部已无内含子等结构,值得说明的是,目前火热的二代测序均是先将RNA反转录组成cDNA再进行测序的。
单拷贝基因与基因家族
单拷贝基因指在基因组中只出现一次,多是编码蛋白质的基因,真核生物中有25%~50%的基因是以单个基因存在的,而其余编码蛋白质的基因以基因家族形式存在;基因家族是来源于同一个祖先,由一个基因通过基因重复而产生两个或更多的拷贝而构成的一组基因,它们在结构和功能上具有明显的相似性,编码相似的蛋白质产物,
假基因
假基因也叫伪基因,他是基因家族在进化过程中形成的无功能的残留物。它与正常基因相似,但丧失正常功能的DNA序列,往往存在于真核生物的多基因家族中,一般情况都不被转录,且没有明确生理意义。
参考资料
