【3.2】整合人类微生物组计划(The Integrative Human Microbiome Project,HMP2)
摘要:
NIH人类微生物组计划(HMP)进行了十年和两个阶段,旨在提供资源,方法和发现,将人类及其微生物群之间的相互作用与健康相关的结果联系起来。最近完成的第二阶段,“综合人类微生物组计划”,包括在以下三个条件下对微生物组和宿主动态变化的研究:怀孕和早产;炎症性肠病; 和影响糖尿病前期个体的压力源。相关研究开始阐明在这些条件下宿主与微生物组相互作用的机制,提供了独特的数据资源(在HMP数据协调中心),并代表了人类微生物组未来多组学研究的范例。
一、前言
尽管“组学时代”加速了生物学研究的各个方面,但其作用在微生物群落和人类微生物组的研究中尤为明显。在第一人类基因组的公布后的18年中,微生物组的研究已经从口腔和肠道的基于培养,到调查生长在人体的所有生态位微生物生物化学的分子概况。流行病学和模型系统已被用于在微生物从孤独症变化和条件之间的关联标识至癌症,并且已经确定了影响例如心脏疾病或移植物抗宿主病的生存期的药物的疗效的微生物和免疫学机制。
对人类微生物组的当代研究也已成为基本生物学和转化学惊喜的来源,暴露了一系列令人信服的新颖发现和悬而未决的问题。每个人似乎携带自己的,这主要是个人,套件的微生物菌株的,它们在生命的早期获得的,环境和群体之间差异,并且可以持续数年或经历相对快速过渡。微生物的多样性在人体不同的生态位中表现出不同的表现。例如,肠道通常具有更大的多样性,但可能与营养不良状态和女性生殖道不良事件的风险有关。微生物组可能会受到炎症性肠病和糖尿病等疾病的干扰,但仍未探索各种与微生物组相关的健康状况以及这些联系的基础。在怀孕或病毒感染等过程中,微生物组的动态性如何?微生物组中的哪些变化代表健康变化的原因而不是影响?个性化微生物组的哪些分子元素可能与健康状况有关,它们如何与免疫系统和新陈代谢等生理过程整合并维持它们的生命?哪些生态因素决定了微生物群移植的成功,为什么它们能够成功地治疗某些个体和病症,但不能成功治疗其他个体和病症?
健康人类微生物组项目的国家机构是最早的大规模举措来解决这些问题的一个子集(图1)。该计划的第一阶段于2007年启动,旨在确定在没有明显疾病的情况下,“健康”微生物群落是否具有共同的要素。这两个基准成人人口的研究具有特定疾病状态的“示范”人群确定了整个人体的微生物组成和酶谱的典型范围(对于某些人群),普遍或特定菌株的代谢功能组合以及某些宿主因素(例如 race or ethnicity)来确定这种差异。识别壁龛生态状态(ecological states of niches )目标人群如阴道的研究,皮肤,和肠道,在许多其他( https://www.hmpdacc.org/health/projectdemos.php )。该HMP(HMP1)的第一阶段从而产生了丰富的社区资源:
- 微生物和社区的核苷酸序列从大量分离物,个人和群体( http://hmpdacc.org );
- 协议来支持可再现的全身性微生物采样和数据生成;
- 和用于微生物分析和流行病学计算方法。

图1 为期十年的NIH人类微生物组计划(HMP)计划分为两个阶段(HMP1和HMP2),开发了参考序列,多组学数据集,计算和统计工具以及分析和临床方案,作为更广泛的研究社区的资源。 HMP1在健康成人受试者的基线研究中着重于从众多身体部位(口腔,鼻,阴道,肠道和皮肤)的微生物群落特征,并包括一系列针对特定疾病或病症的示范项目。在代表性微生物组相关疾病的三个纵向队列研究中,HMP2扩展了宿主和微生物组生物学特性的库:妊娠和早产(孕妇的阴道微生物组),炎症性肠病(肠道微生物组)和糖尿病前期(肠道和微生物)。鼻微生物群)。这些研究通过随时间推移对多种测量类型进行多组学分析来跟踪这些条件的动态变化,包括微生物群落组成,病毒学,代谢组学谱,宿主和微生物组的基因表达和蛋白质谱以及宿主特异性特性(例如遗传,表观基因组,抗体和细胞因子概况,以及其他研究特定特征。来自HMP1和HMP2的所有序列和多组数据,临床信息以及工具都存放在HMP数据协调中心(DCC)或引用的公共或受控访问存储库中,以作为研究社区的中央资源。
HMP1的主要发现之一是,单独的微生物组的分类学组成通常与宿主表型之间没有很好的相关性-倾向于通过普遍的微生物分子功能或个性化的菌株特异性组成来更好地预测。这一发现为HMP第二阶段(集成HMP(iHMP或HMP2))的开发奠定了基础,旨在探索宿主-微生物组之间的相互作用,包括免疫力,新陈代谢和动态分子活性,以便随着时间的推移更全面地了解宿主-微生物之间的相互作用。该多基因组计划旨在扩展微生物组研究社区可用的资源基础,开始以机械方式解决宿主与微生物组之间的关系,并解决上述问题。因此,鼓励在HMP2内以疾病为目标的项目使用多种补充方法,以纵向评估人类和微生物活动的机制,并为今后的工作提供方案,数据和生物标本。这些项目包括三项研究,这些研究追踪了已知微生物组相互作用条件下人类健康和疾病的动态,因此可以直接解决重要的健康后果,同时还可以作为研究团体广泛关注的“典型”微生物组相关状况的模型。这些包括怀孕和早产(PTB, pregnancy and preterm birth );炎症性肠病(IBD, inflammatory bowel diseases); 和影响糖尿病前期个体的压力源。这些研究现已进入完成的第一阶,一起提供了丰富的信息和见解的不仅是微生物动态,而且有关的人类宿主反应和微生物的相互关系。迄今为止,在 https://www.nature.com/collections/fiabfcjbfj 上收集了20余份手稿,描述了其中的一些结果,它们共同提供了丰富的多组数据资源,可供以后的工作( http://www.ihmpdcc.org )。
二、阴道微生物组,怀孕和早产 The vaginal microbiome, pregnancy and preterm birth
早产会对新生婴儿造成毁灭性后果,包括死亡和长期残疾。在美国,约有10%的婴儿是早产,而在资源较少的国家,这一比例甚至更高。环境因素,包括女性生殖道的微生物组,是导致早产的重要因素。值得注意的是,这些因素对非洲裔妇女的影响更大,非洲裔妇女也承担着最高的PTB负担。近几十年来,婴儿死亡率有所降低,但是PTB的发病率并未下降,预测个人PTB风险的进展停滞了。怀孕期间,孕妇的免疫系统维持促炎和抗炎效应子之间的微妙平衡,而PTB的病因包括母体-胎儿耐受力下降,血管疾病,压力,宫颈机能不全,胎膜早破和羊膜内感染。微生物升入子宫被认为通过破坏母体的免疫平衡,导致自发性早产和/或通过释放损害胎膜和胎膜完整性的微生物产物(例如胶原酶,蛋白酶或毒素)导致膜过早破裂从而PTB。。
作为HMP2的一部分,Multi-Omic Microbiome Study:Pregnancy Initiative(MOMS-PI)研究小组对孕妇的微生物群进行了表征,以评估其对PTB风险的影响(图2)。)。该项目纵向跟踪了1,527名妇女直至怀孕的整个过程,并收集了206,437个标本,包括孕妇阴道,颊,直肠,皮肤和鼻孔拭子,血液,尿液和出生产品,以及婴儿脐带血,脐带血和胎粪,颊,皮肤和直肠拭子。这些标本的子集进行了16S rRNA基因分类学分析,宏基因组和元转录组测序,细胞因子谱分析,脂质组学分析和细菌基因组分析。MOMS-PI团队分析了597例怀孕的12,039份样本,以研究微生物组的动力学及其与妊娠期间导致PTB 50的宿主之间的相互作用。
MOMS-PI项目纵向进行了1,527例妊娠,涉及206,437份生物标本的收集,用于分析宿主和微生物因素(16S扩增子,宏基因组和元转录组测序;细胞因子谱;代谢组学;蛋白质组学;基因组学和微生物分离培养)。对大约600例怀孕进行了深入分析,以评估导致早产的特征。该分析确定了宿主(例如细胞因子)和微生物(例如生态和特定菌株)因素。随着怀孕的进行,全身性雌二醇水平发生可预测的变化,子宫和阴道环境也会发生各种变化。子宫在妊娠中期从早期的促炎状态转变为抗炎状态,然后在分娩开始之前回到促炎状态。同时,阴道管腔微生物组的特定变化可能与早产有关,可能是通过涉及微生物从阴道行进到子宫的机制引起的。该图概述了怀孕期间阴道粘膜生态系统和子宫的纵向变化。
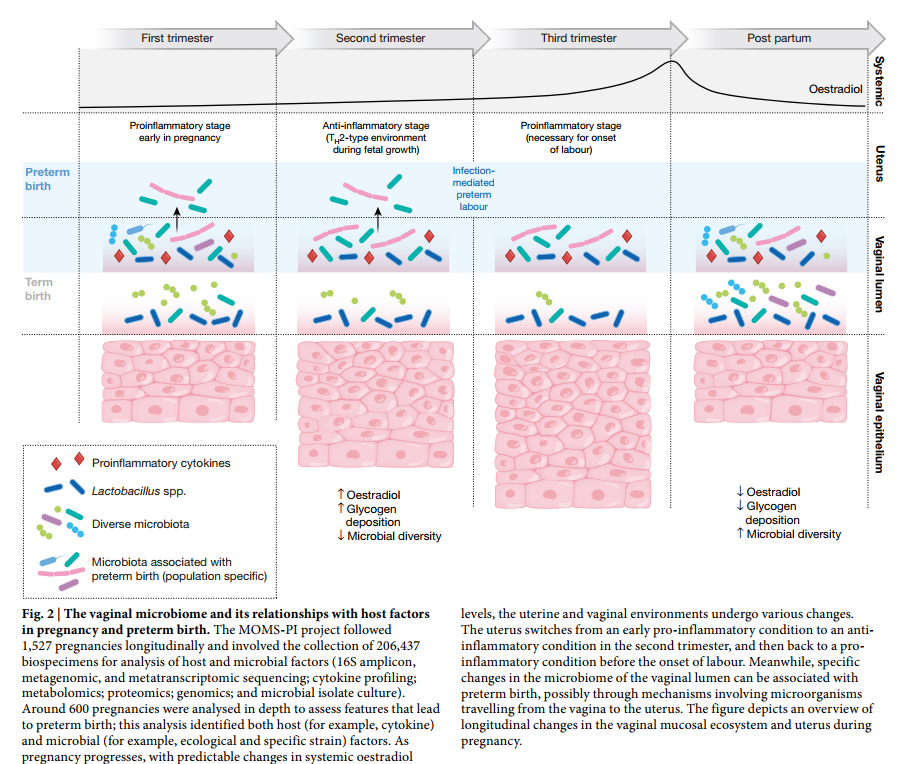
这些多组学研究确定了与足月妊娠相关的阴道微生物组的时间变化。经常怀孕的妇女通常会在生态学上更为复杂的阴道微生物组中怀孕,通常在孕中期开始趋向于以乳杆菌(Lactobacillus)为主的微生物组更为均一。有趣的是,这种趋势在社会经济地位较低的非洲裔女性中最为明显。尽管整个MOMS-PI队列的人口统计学差异很大,但大多数在妊娠少于37周时经历自发性PTB的女性是非洲人。MOMS-PI团队( http://vmc.vcu.edu/momspi )还确定了:
- 在妊娠少于37周时自发早产的妇女发生PTB的风险较高[ 50]。
- PTB妇女不太可能表现出由主导的阴道菌群卷曲乳杆( Lactobacillus crispatus),如以前在其他人群中报道的,并更可能呈现出增加的几个类群包括 Sneathia amnii, Prevotella-related clades, a Lachnospiraceae taxon known as BVAB1, and a Saccharibacteria bacterium known as TM7-H1。值得注意的是,这些分类群也与维生素D含量低有关,表明阴道微生物组可能介导PTB风险与维生素D缺乏症之间的联系。
- PTB的特征也反映在宏基因组学和元转录组学测量中,并且阴道促炎细胞因子(包括IL-1β,IL-6,MIP-1β和eotaxin-1)与PTB相关的分类单元呈正相关。为便于将来可能的干预,经历PTB的母亲的阴道微生物组与怀孕早期的对照母亲的阴道微生物组最为不同,并且使用从24周前收集的样本中的阴道微生物组谱资料预测PTB风险的初步模型最为敏感和特异性妊娠
MOMS-PI研究小组确定了在至少某些自发性PTB病例中,阴道微生物群,宿主反应和妊娠结局之间有趣的关联,这些关联与从阴道升起的微生物的侵袭一致。作为必不可少的下一步,必须通过统一的大规模研究充分探讨种族和人口统计学背景对妊娠阴道微生物组与妊娠结局的影响。显然,PTB具有复杂的病因学。应该探索胎儿和母亲遗传学和表观遗传学的相对贡献,特别是与先天免疫系统的遗传变异有关的贡献。大规模的协调研究将允许使用阴道微生物组谱,基因和产前(胎儿)基因筛查的特征,诸如细胞因子和代谢产物的生物标志物以及包括孕妇在内的经典危险标志物的关键临床特征来开发针对特定人群的风险评估算法年龄,体重指数,怀孕史(包括PTB病史),宫颈长度以及压力和其他环境暴露量度。通过添加来自微生物组,其他环境因素和多组学输入的新数据,新算法有望提高我们预测怀孕初期PTB风险的能力,
三、肠道微生物组和炎症性肠病
在胃肠道疾病中对肠道微生物组的研究有着悠久而详尽的历史,特别是在复杂的慢性疾病(如炎症性肠病(IBD, inflammatory bowel diseases )中。IBD,包括克罗恩氏病和溃疡性结肠炎,在全球范围内影响着数以百万计的人,在过去的50年或更长的时间内发病率不断上升,同时与多种因素同时发生,如西化,城市化,饮食结构变化,抗菌素暴露以及可能影响宿主的更多因素–微生物组稳态。微生物组早已牵连IBD,作为潜在的致病或风险因子,以解释治疗反应的异质性(也就是说,一些个体对相对良性的氨基水杨酸酯或皮质类固醇反应良好,而其他个体甚至在手术干预后仍会出现严重的炎症反应),或作为治疗干预的新观点(例如,通过粪便菌群的移植))。尽管已使用基因组学技术来鉴定功能一致的微生物反应,这有助于解释肠道微生物组在疾病期间作为肠道促炎性反馈回路的一部分的作用,但一些微生物菌株已被证明是IBD特定的,尚无特定的微生物,分子和免疫相互作用的综合模型来解释这种疾病的发作和动态进展。
因此,为了更好地描述疾病期间宿主-微生物组失调的机制,炎症性肠病多组学数据库(IBDMDB, Inflammatory Bowel Disease Multi’omics Database )项目在一年的过程中追踪了来自五个临床中心的132个人作为HMP2的一部分(图3)。通过分析1,785份粪便样品(每两周自行收集并通过邮件发送),651份肠活检(基线时通过结肠镜检查收集)和529份季度血样,生成了微生物和宿主活动的完整纵向分子图。从相同的样本集,包括粪便宏基因组中产生可能的,多个分子概况的程度,metatranscriptomes ,metaproteomes,viromes,代谢组,宿主外显子组,表观基因组,转录组和血清学特征等,可随时间观察多种宿主和微生物分子及临床活动的同时变化。从研究中,关于其基础设施的进一步的信息,和协议和结果都原始和处理种数据产品都可以通过IBDMDB数据门户( http://ibdmdb.org )中,从HMP2数据协调中心(DCC; http://ihmpdcc.org ),以及随附的手稿。
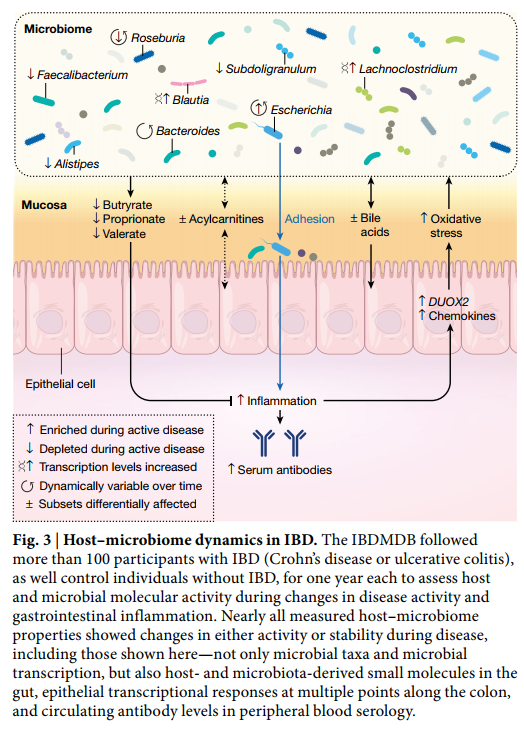
这项独特的研究设计使IBDMDB能够识别微生物组的各种差异,并在疾病过程中随着时间的推移产生宿主免疫反应。事实上,这些动态变化要大得多数量级比分别为临床表型间的横截面的差异,已经强调了以前的研究。这部分归因于该队列的前瞻性,该队列招募了克罗恩氏病或溃疡性结肠炎患者,疾病处于活动期和静止期,这表明IBD患者的微生物组成经常恢复为更像对照的“基线”水平疾病不活跃时的配置。通过确定与基线最不相同的肠道微生物构型(无论具体疾病状况如何),该研究定义了一种营养不良评分,该评分指出了高度不同的微生物成分,这些成分具有整体炎症反应的许多共同特征(例如,对氧化的耐受性)压力)。这种菌群异常并不是微生物对炎症的反应所独有的,并且与其他宿主和生化改变有关,指出了治疗IBD系统失调的新潜在方向。这些包括酰基肉碱池和胆汁酸的大幅变化,血清抗体水平的提高以及几种微生物物种的转录变化。从活检组织中同时进行的转录组学和16S扩增子黏膜群落特征分析还确定了可能能够塑造微生物群落的潜在宿主因子,特别是几种趋化因子,强调这些因子参与疾病活动期间潜在的相互作用失调。
该研究的纵向多组学特征进一步使研究人员能够表征疾病期间宿主与微生物组相互作用的稳定性和动力学,特别是突出显示了在IBD参与者中,社区状态和免疫反应比在健康人中明显不稳定的方式。在许多情况下,IBD参与者的微生物组仅在数周内就发生了完全变化(以与同一受试者早期样本的最大Bray-Curtis相似性衡量),而这种变化在没有IBD的个体中很少见。这些从一个时间点到下一个时间点的大规模转变的主要微生物来源,在很大程度上反映了在菌群失调中观察到的差异,而且这种转变经常标志着菌群失调时期的进入或退出。最后,这项研究是长期的 互补的分子测量可在IBD期间构建由2,900多种重要的宿主,微生物细胞和分子相互作用物组成的网络,范围从特定的微生物分类群到人类转录本和小分子代谢物。这个机械协会网络确定了IBD发生变化的关键几个关键组成部分,突出了辛酰基肉碱,几种脂质和短链脂肪酸,分类单元Faecalibacterium,Subdoligranulum,Roseburia,Alistipes和Escherichia,一些处于宏基因组和超转录组水平,并且是白介素的宿主调节因子。诸如此类的机械关联网络可能为解开相互作用的复杂系统提供了关键,而复杂的相互作用系统会导致IBD和其他系统性微生物组相关免疫疾病的慢性炎症。
四、糖尿病前期的多组学分析 Multi-omics profiling in prediabetes
2型糖尿病(T2D,Type 2 diabetes mellitus)影响超过10%的美国成年人口,另有30%的人显示出该疾病的早期征兆(称为糖尿病前期)。另外中有70%会在一生中患上糖尿病。 T2D的特征在于复杂的宿主-微生物相互作用,但对糖尿病前期的系统性改变,其对生物过程的影响或向全面成熟的T2D的关键转变的了解却很少。糖尿病前期和T2D通常与糖尿病抵抗相关,因此对糖尿病糖尿病前期或糖尿病抵抗的个体的研究提供了独特的机会来研究糖尿病的早期阶段。为了及时了解糖尿病前期个体中储存和微生物分子的堆叠和同时分布,这是必不可少的不可少的,充分了解糖尿病前期和/或抵抗抵抗患者中毒的分子途径以及这些状况如何影响生物学响应对环境挑战(例如,病毒感染)和T2D的发作。
为了更好地了解T2D,作为iHMP的一部分,个人Omics项目(IPOP)在大约每季度的季度健康,呼吸道病毒感染(RVI)和其他扰动期间跟踪了106名健康和糖尿病前个体(图4)。在这种扰动中,有23个人的一个子集进行了定向减肥,然后又进行了减肥。总计分析了所有参与者的1,092个收藏夹。对于每次访问,都对血液进行了宿主分子组学谱分析,并采集了两种类型的样品(鼻拭子和粪便)进行微生物谱分析。每个参与者的外显子组插入一次。否则,每次访问都从穿过血单核细胞中提取13379个转录本,从血浆中提取722种代谢产物和302种蛋白质,从血清中提取62种细胞因子和生长因子。使用16S rRNA扩增子分析了成千上万的肠仍通过51项临床实验室测试对所有访视进行了深入分析。如此,由于专注于T2D,因此进行了许多葡萄糖失调测试,包括对空腹血糖和血红蛋白A1C水平的测量,
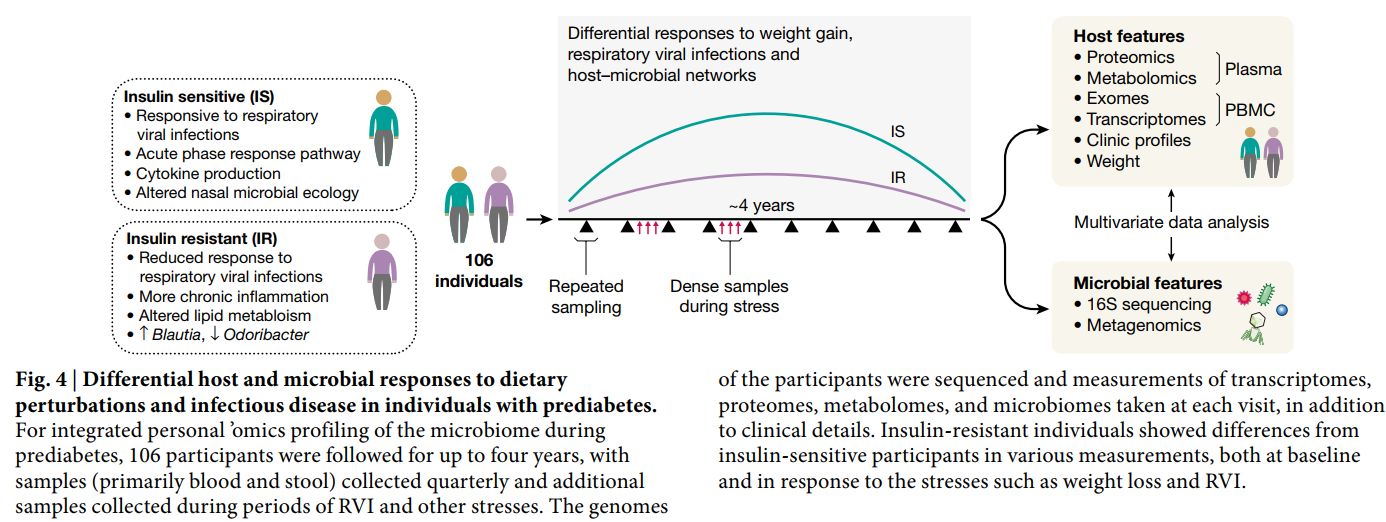
即使在很长一段时间内,个体内的基线测量值通常也是稳定的,只有一些分析物随时间发生显着变化。但是,许多分析物,例如临床实验室测量结果,细胞因子谱和肠道微生物分类群(大多是低丰度的那些)在个体之间变化很大。最终具有胰岛素抵抗性的参与者在基线时与那些最终具有胰岛素敏感性的参与者在分子和微生物上有明显的区别,因此研究人员设计了分析物测试以区分它们。值得注意的是,经历RVI或体重变化的个体在这些扰动期间显示出数千种特定的分子和微生物变化,而胰岛素抵抗和胰岛素敏感的个体对扰动的反应也非常不同。例如,在RVI期间,与胰岛素敏感的参与者(例如在Lachnospiraceae和Lasnospiraceae中)相比,胰岛素抵抗的参与者表现出明显降低和延迟的炎症反应(例如,急性期反应和IL-1信号传导)和肠道微生物变化的改变。菊科但不包括杆菌)。因此,胰岛素抵抗患者中鼻腔菌群的变化较少,胰岛素敏感性但非胰岛素抵抗者中RVI期间鼻微生物的丰富度和多样性均降低。此外,对数千个分析分子之间的全球关联分析表明,胰岛素抵抗性个体中的特异性关联不同于对胰岛素敏感的参与者,反之亦然,这表明两组中宿主-微生物组相互作用的方式不同51。
该研究的另一个重要目标是评估如何使用宿主-微生物组多组学和相关的新兴技术更好地管理患者的健康。我们发现随着时间的推移对每个人进行数百万次测量可以早期发现潜在的疾病状态。其中包括早期检测T2D,其在参与者之间发展不同,并且可以通过各种检测方法更好地检测到。例如,有些人首先在空腹血糖测试中显示出在糖尿病范围内的测量值,而另一些人则在血红蛋白A1c测试,口服葡萄糖耐量测试或什至连续血糖监测中进行了测量。这些结果,以及随时间变化的葡萄糖失调的详细特征,说明了T2D发育的异质性。总体而言,这些数据导致了除T2D以外的多种疾病的微生物相关的,可在临床上应用的健康发现,包括代谢性疾病,心血管疾病,血液学或肿瘤学状况以及其他领域;这些迹象通常在症状发作之前就出现,表明使用包括微生物组在内的大数据来更好地管理人类健康的力量。
五、HMP2的资源
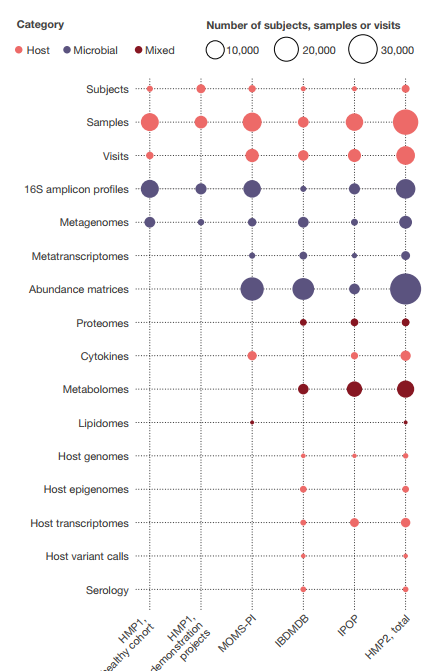
HMP1和HMP2阶段共产生了总计42 TB的多组数据,这些数据由DCC在 http://ihmpdcc.org 以及公共和/或受控访问存储库(如序列读取档案(SRA; https://www.ncbi.nlm.nih.gov/sra ),基因型和表型数据库(dbGaP; https://www.ncbi.nlm.nih.gov/gap/ ),代谢组学工作台( https://www.metabolomicsworkbench.org/ )等(图5)。DCC上的所有数据均可无限制使用,在机构审查委员会(IRB)允许的情况下,也可共享项目元数据的子集,而其他受限制的数据(例如,人类基因组序列和受保护的元数据)可通过以下方式进行受控访问: dbGaP(项目PRJNA398089,PRJNA430481,PRJNA430482,PRJNA326441,phs001719,phs000256,phs001626,phs001523等)。HMP各阶段产生的正式数据模型和关联的实体关系模式可从 https://github.com/ihmpdcc/osdf-schemas 免费获得。DCC网站允许用户从数千个具有相关元数据的样本中查找,查询,搜索,可视化和下载数据。一旦用户识别出一组感兴趣的文件,条件,主题或表型,他或她便可以将此集合添加到购物车中以进行进一步的操作。然后可以直接下载文件以在用户的本地站点或云中使用。因此,HMP DCC的设计与NIH的既定目标保持一致,以使从NIH资金中产生的所有数据都可发现,可访问,可互操作和可重复使用88。用户对Web资源的访问率一直很高,每月有9,000-12,000个用户会话,并且在此处发布这些资源后,预计吞吐量会不断提高,证明了这些努力的成功。
六、互补的宿主-微生物组相互作用
尽管三项HMP2研究均揭示了各自健康和疾病领域的新生物学,但它们之间常见的宿主-微生物组免疫和生态学特征令人惊讶。 shot弹枪宏基因组学,非靶向代谢组学和免疫分析的组合特别有效,因为在所有项目中,分子测量的这一子集都倾向于有效地捕获与疾病有关的可解释的宿主和微生物特性。相反,在如此小的种群中,遗传变异通常很难与微生物组关联,这对于随着时间的推移深入剖析多组学是必不可少的,并且我们预计将宿主测序整合到较大的横断面调查中会更加有用。与大多数微生物组研究一样,另一个值得注意的特性是,个体,群体或表型内部发生的变化通常比个体之间的基线变化小得多。在与微生物组相关的时间尺度上尤其如此,对于这种时间尺度,必须采取最快至几天至几周的重复措施才能捕获最具体的宿主与微生物组的相互作用。因此,与健康相关的微生物组相互作用可以在个体之间以极其多样化的方式表现出来,从而使大规模的人口调查与受试者内部的纵向概况相结合,对于理解微生物相关疾病的机制至关重要。
结果,在三项研究中的每项研究中,宿主与微生物组相互作用的其他方面都高度局限且具有特定的主题。在所有这三种情况下,当发生变化时捕获的微生物变化和相关的宿主反应最强,并且通常在起源组织内。因此,从这些研究和其他研究中可以清楚地看出,宿主与微生物组的相互作用既具有局部作用,又具有全身作用。从宿主或微生物一侧引发的强烈局部扰动可诱导随后的时空响应,该响应可能随时间和/或在其他组织中持续发生,大概是通过循环中的小分子在空间上和/或在时间上通过基因调控或微生物的生长所携带的信号,并涉及具有宿主和微生物成分的调节电路。因此,持续的协调努力以测量每种疾病所涉及的多种宿主和微生物特性,对于开发针对微生物组的相关疾病的针对性(必要时)个性化疗法以及揭示控制宿主与微生物组相互作用的一般原则至关重要。在所有研究中均未测量到的其他动态相互作用,例如一个人在出生时的首次微生物暴露以及随后的免疫发育,也可能代表了基线微生物组个性化的关键因素,并有助于根据发生数年或以上的事件来解释与疾病相关的动力学。。
七、微生物组学的下一步
NIH HMP项目的集体结果以及许多其他研究表明,微生物组是人类生物学的组成部分,对健康和福祉起着重要作用。随着时间的流逝,个体间的变异性和高度多样的宿主-微生物组反应推动了使用多种互补的纵向测量方法进行人群微生物组研究的新方法的发展,并强调需要用机理模型对此类研究进行跟踪以验证病因协会。HMP计划本身的成功结束,留下了多代受过训练的人类微生物组研究人员的科学遗产。为最终的社区提供了丰富的数据,分析和生物样本资源;89==。NIH中心和研究所之间正在协调微生物学(人类和其他方面)的资金筹集( https://www.niaid.nih.gov/research/trans-nih-microbiome-working-group );其他美国政府机构,包括国家科学基金会,环境保护局,能源部,国家标准与技术研究院,农业部,国家海洋与大气管理局,国家航空航天局和国防部( https://commonfund.nih.gov/hmp/programhighlights ); 慈善组织,包括比尔和梅琳达·盖茨基金会,三月的一角钱,巴勒斯惠康基金会,斯隆基金会,凯克基金会,青少年糖尿病研究基金会,克罗恩氏和结肠炎基金会等;以及行业和公私合作伙伴关系。此外,随着启动针对个性化医学各个方面的复杂的全球项目,现在很明显,将关注人类微生物组作用的成分包括在内是有益的。
与任何大型研究一样,HMP2提出的新问题多于已回答的问题。即使从这三项研究和人群中纳入的各种测量类型来看,微生物组的基线个体间差异及其随时间的动态变化的病因学也并不明显。许多免疫和生化反应似乎与一种或几种个体宿主所特有的特定菌株有关,但尚不清楚此类菌株是否足够或必需用于其相关疾病表型。确定了几种机制,通过这些机制可以将肠道中的信号传递至全身性疾病,例如糖尿病,但是可能不会通过它们传播的特定小分子或免疫细胞亚群-尤其是在尚未进行如此详细研究的其他健康状况中。最后,每项HMP2研究都必须在受地理和基因限制的人群中进行,并且早期生命事件,传染病暴露或饮食方面的全球差异可能会改变微生物组动力学对人类疾病的影响。现在,与人类有关的微生物学已经明显地从传染病和胃肠道疾病扩展到了几十年前难以想象的领域,包括新陈代谢,肿瘤,母婴健康以及中枢神经系统功能。随着NIH HMP的结束,很明显,其结果揭示了许多新的研究方法和技术,可用于未来的研究,
参考资料
- Published: 29 May 2019. The Integrative Human Microbiome Project. https://www.nature.com/articles/s41586-019-1238-8
