【6.4】DeepCRISPR
2018年6月26号,同济大学刘琦研究组,黄德双研究组及阿斯利康Jia Wei合作在Genome Biology在线发表题为“DeepCRISPR: optimized CRISPR guide RNA design by deep learning”的研究论文,该论文开发了一套新的方法,简称为DeepCRISPR,它基于精心设计的用于模型训练和预测的混合深度神经网络。
有效应用CRISPR系统的主要挑战是准确预测单个向导RNA(sgRNA)的靶标敲除效率和脱靶情况,这将有助于以高灵敏度和特异性进行sgRNA的优化设计。 在这里,我们介绍了DeepCRISPR,这是一个全面的计算平台,可通过深度学习将sgRNA的on-target and off-target 统一到一个框架中,超越了现有的最新计算机工具。 此外,DeepCRISPR以数据驱动方式,完全自动化序列和表观遗传学特征的鉴定,这些序列和表观遗传学特征可能会影响sgRNA敲除功效。 DeepCRISPR可从 http://www.deepcrispr.net/ 获得。
一、前言
基于CRISPR的基因敲除已广泛应用于各种细胞类型和生物中。在该系统中,单向导RNA(sgRNA)将Cas9蛋白引导至特定的基因组靶标。识别和切割是通过sgRNA中的20个核苷酸(nt)序列与基因组靶标,在其3’末端的原间隔子相邻基序(PAM)的目标上游]。广泛的研究还表明,可以容忍多个错配以及DNA或RNA凸起,从而导致切割意想不到的基因组位点(称为脱靶,off-targets)[2]。这种CRISPR-Cas9核酸内切酶系统允许以核苷酸分辨率编辑基因组[3,4],而其有效应用的主要挑战是事先准确预测sgRNA的靶标敲除功效和off-target(OT)。准确的预测将通过最大化sgRNA的靶向作用(高灵敏度)和最小化其脱靶作用(高特异性)来促进sgRNA的优化设计[1,2,5-7]。
已经开发了各种sgRNA设计规则和工具来进行sgRNA的靶标鉴定和功效预测。这些方法分为三种类型:
- 基于比对,其中sgRNA仅通过定位PAM与给定的基因组进行比对(CasFinder [8]等);
- 假设驱动( hypothesis-driven),通过考虑基因组背景因素(E-CRISP [9],CRISPR [6],CHOPCHOP [10],GuideScan [11]等)的经验,对sgRNA敲除效率进行经验评分;
- 基于学习的方法,其中通过考虑不同特征从训练模型中预测sgRNA敲除效率(sgRNA Designer [2],SSC [5],sgRNA Scorer [12、13],CRISPRscan [14]等) 。
一项基准研究表明,后两种类型的工具通常比基于比对的工具表现更好,尽管在不同的细胞类型上预测并不能很好地扩展[15,16]。需要新颖的计算方法,以及对sgRNA敲除效率有影响的DNA序列和表观遗传学特征的全面探索[7,16]。
脱靶被证明在CRISPR系统中发生[2,6,17]。尽管在特定位点进行sgRNA引导的Cas9切割不一定会导致功能序列(例如框内移位突变)[18],但如何准确,定量地检测或预测脱靶切割位点仍然存在一个重要的问题,仍然具有挑战性[19]。
- 大多数现有工具使用具有不同核苷酸错配的简单序列比对来彻底搜索脱靶位点[20]。
- 一些工具通过设计脱靶得分(例如,CFD得分[2],MIT得分[6、16、21]等)来预测错配基因座的切割效率。 他们的预测结果与各种技术生成的数据进行了比较,例如:sgRNA脱靶裂解检测技术(如GUIDE-seq ,Digenome-seq ),高通量全基因组易位测序(HTGTS),直接原位中断标记测序(BLESS)[26]和整合缺陷型慢病毒载体捕获(IDLV)[27]。
这些都是本质上基于假设的方法,它们使用凭经验定义的脱靶标准来识别脱靶位点。需要对整个基因组脱靶谱进行有效的基于学习的预测。
目前,建立用于sgRNA功效预测的学习模型面临几个障碍:
- 数据异质性问题,其中来自不同细胞类型和实验平台的数据需要有效整合。
- 数据稀疏性问题,其中标记的样本量(即具有已知功效的sgRNA的数量)相对较小,而且收集起来的实验费用昂贵-标记的数据不足会使当前的学习模型效率低下;
- 脱靶位点预测中的数据失衡问题—在所有可能的核苷酸错配位点中,全基因组脱靶检测技术识别的真正脱靶切割位点的数量很小;
- 影响sgRNA功效的前导序列和表观遗传学特征尚不清楚,尚待进一步探索[5]。
最近,有几项研究尝试了复杂的学习模型来进行on-target or off-target的预测[28-30],但是它们都没有彻底解决这些问题。在我们的研究中,我们提出了一个新颖而强大的深度学习框架[31-33],可同时预测sgRNA的靶标敲除功效和全基因组的off-targetcleavage profile,可与现有的工具竞争。我们的方法称为DeepCRISPR,基于精心设计的混合深度神经网络,用于模型训练和预测。据我们所知,这是最全面的计算平台,可用于通过深度学习将目标上和目标外的站点预测统一为一个框架。我们应用了深度无监督的表示学习技术[32,34],使用一整套全基因组范围的未标记的sgRNA自动学习sgRNA的基本表示。使用现有的标记sgRNA,通过监督的深度神经网络进一步调整学习的模型。我们指出,大量未标记的sgRNA的预训练可用于增强模型预测,这在传统sgRNA功效预测中从未进行过研究。
Deep-CRISPR共同具有以下优势,解决了上述挑战:
-
通过考虑不同细胞类型中的表观遗传信息,它在统一的特征空间中代表了来自不同细胞类型的不同DNA区域,并整合了来自不同实验的数据-元素和单元格类型。尽管DeepCRISPR在有限的细胞类型数据上受过训练,但我们验证了它在适应新细胞类型时通常具有良好的预测能力。
-
它从数十亿个全基因组未标记的sgRNA中学习,以自动生成“父网络”,从而同时针对sgRNA的靶标和靶标设计生成高级特征表示。通过这种方式,DeepCRISPR通过考虑来自这些区域的全基因组sgRNA序列的无监督预训练,优化了编码区和非编码区的sgRNA设计。比较了有和没有预训练的模型,并验证了无监督预训练的优越性。
-
它应用了一种特殊的数据增强技术来生成具有生物学意义的标记的新型sgRNA,从而增加了sgRNA靶上位点预测中标记的训练大小。我们进一步证明,这种数据增强确实提高了预测性能,并使训练模型更强大。
-
它进一步使用标记的sgRNA数据微调父网络,这有助于提高对有限标记样本的预测性能。
-
它结合了有效的自举采样算法和训练程序,大大减轻了脱靶站点预测中的数据不平衡问题。
-
最后,它使序列和表观遗传特征的识别完全自动化。该模型使用有限的训练样本了解哪些功能对优化sgRNA设计很重要。鉴定出的特征可用于优化的sgRNA设计。这有助于以更有效的数据驱动方式解密CRISPR的 on-target and off-target机制。
DeepCRISPR可从 http://www.deepcrispr.net /获得。 命令行代码也可从 https://github.com/bm2-lab/DeepCRISPR 和 https://zeno-do.org/record/1246320 获得。 Deep-CRISPR的当前版本专注于人类SpCas9的基于NGG的常规sgRNA设计。 它可以轻松扩展到其他Cas9物种或变体和其他物种。 将其在on-target and off-target site预测性能与可用的最新工具进行了比较。
二、结果
2.1 训练DeepCRISPR进行sgRNA on-target and off-target预测
2.1.1 sgRNA表示的深度无监督学习
DeepCRISPR的第一个输入是完整的20 bp sgRNA序列集,整个人类基因组中均带有NGG PAM。我们用NGG PAM从人类编码区和非编码区提取了所有sgRNA序列。这些数据说明了来自大约13种人类细胞的具有不同表观遗传信息的6.8亿个sgRNA序列(请参见“编码具有基因组和表观遗传特征的sgRNA”部分)。它们可作为大规模的未标记sgRNA数据源,用于以下预训练程序,以得出sgRNA的有效特征表示。整个数据收集和预处理是通过使用基于SPARK的具有图形处理单元(GPU)加速的大规模数据处理体系结构来实现的。每个sgRNA最初都用其序列和表观遗传信息编码(请参见“编码具有基因组和表观遗传学特征的sgRNA”部分)。然后,利用这些未标记的sgRNA序列,我们使用深度无监督的表示学习策略来训练基于深度卷积表示神经网络(DCDNN)的自动编码器[35],以自动学习无监督的sgRNA的潜在有意义的表示形式方式[34](图1c;请参见“用于表示学习的基于DCDNN的自动编码器”部分)。这种降噪策略有助于训练自动编码器,以鲁棒性地容忍大量sgRNA数据中的噪声。使用自动编码器的直观原理是,可以使用带有编码和解码的未标记数据来学习有效的特征表示。这样学习到的特征表示将适合以下模型构建。在此步骤中训练的网络称为无监督,预训练的父级网络,以进行进一步分析。
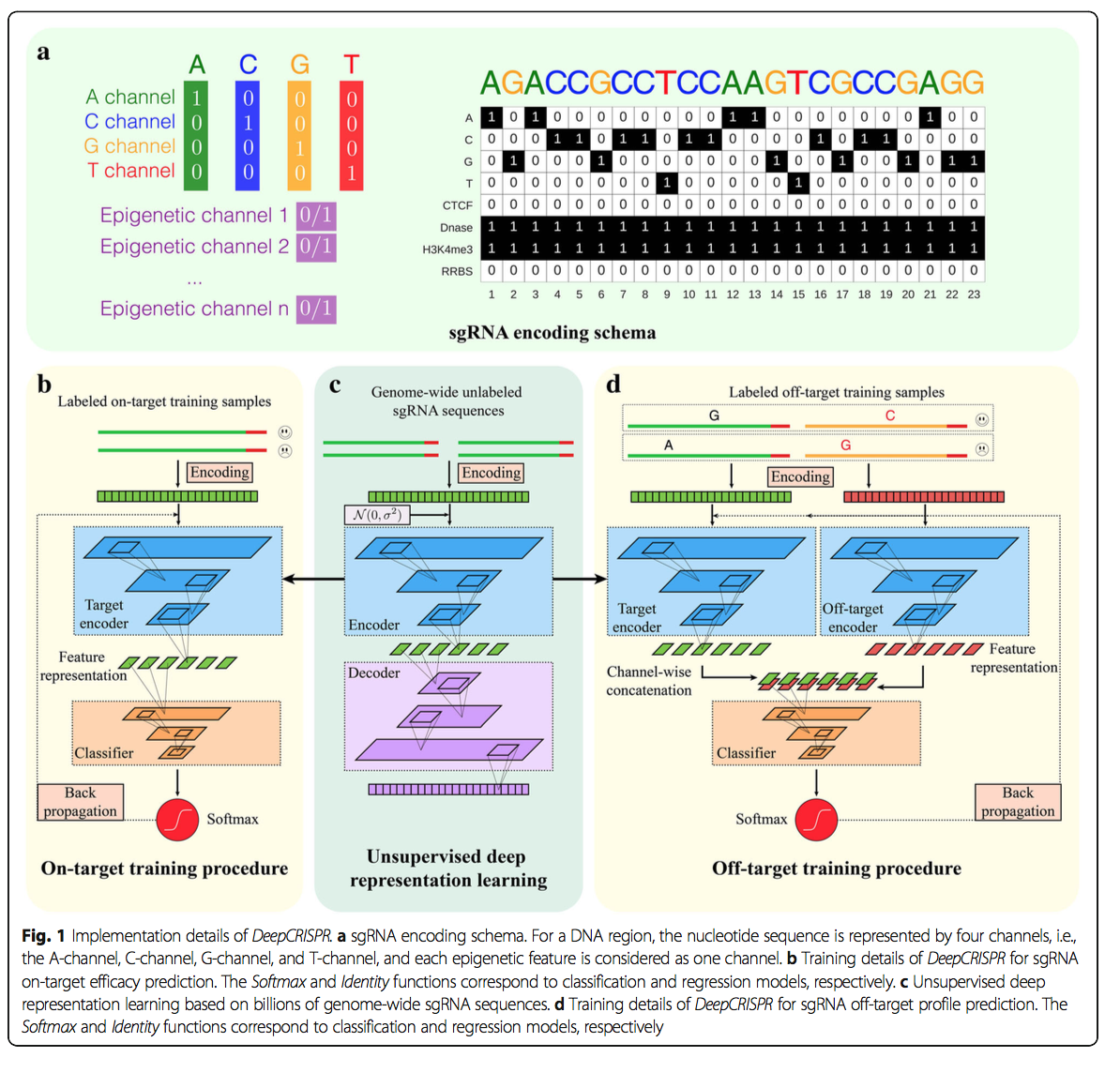
2.1.2 用于sgRNA击靶功效预测的微调混合深度神经网络
A fine-tuned hybrid deep neural network for sgRNA on-target knockout efficacy prediction
接下来,我们生成了一个用于sgRNA击靶功效预测的混合深度神经网络,包括两个部分。第一部分是以前的基于DCDNN的预训练网络(父网络),其输出用作卷积神经网络(CNN;图1b,c;请参见带有预训练的CNN模型)的输入。基础微调”部分)。然后根据标记数据训练整个混合神经网络,即收集的具有已知击中效率的sgRNA。训练过程不仅了解了基于CNN的网络的权重,而且还微调了父网络的权重。因此,该策略使用有限的标记数据来调整原始的预训练网络权重,并有望提高预测准确性(图1b,c;请参阅“基于预训练的CNN模型进行微调”部分)。在我们的研究中,标记的sgRNA数据集包含约20万个具有已知敲除效率的sgRNA。该数据集是从1071个基因中的约15,000个sgRNA产生的,具有已知的敲除效率,且采用数据增强的方式(请参见“目标数据源”部分),类似于图像数据处理所用的方法(“针对目标数据集的数据扩充”部分)。整个混合深度神经网络的最终调整权重用于预测新sgRNA的靶向敲除效果。另外,为了获得对DeepCRISPR的严格评估,针对目标预测的分类和回归模型均已建立,以进行全面比较。
2.1.3 通过重用父网络扩展sgRNA脱靶位点预测模型
我们还通过重用预先训练的父网络扩展了用于sgRNA脱靶谱预测的混合神经网络(图1c,d)。首先,我们将给定的sgRNA及其一个可能的脱靶基因座作为样品对,并将这些样品对作为脱靶训练样品。样本对分为两部分,其中一部分代表给定sgRNA的编码,另一部分代表其可能的脱靶基因座的编码(图1c;请参见“具有基因组和表观遗传学特征的sgRNA编码”)部分)。通过将原始sgRNA序列和错配序列作为一个整体来考虑,这种由两部分组成的编码可以准确,全面地表示脱靶样品。在训练过程中,将sgRNA脱靶样本的每个部分装入预先训练的基于DCDNN的网络(即父网络)中,以学习特征表示。接下来,针对下一个CNN分类器,将该父网络的输出按通道合并在一起,类似于目标站点预测(图1c)。在我们的研究中,基于收集的标记的sgRNA脱靶数据集(包含约160,000个样本)对完整的混合神经网络进行了训练(请参见“脱靶数据源”部分)。该混合网络的批量训练中集成了有效的自举算法,从而减轻了脱靶站点预测中的数据不平衡问题(图5;请参阅“将引导程序集成到深度神经网络的批处理中以解决数据不平衡问题”部分)。与目标站点预测相似,训练过程不仅学习了CNN网络的权重,还调整了父网络的权重,从而导致两个不同的sgRNA的两个“婴儿网络”关闭-目标样本。两个婴儿网络以及CNN网络的最终调整权重用于预测给定sgRNA的脱靶谱。同样,为了获得对DeepCRISPR的严格评估,建立了脱靶预测的分类和回归模型以进行全面比较。
2.2 DeepCRISPR与最新sgRNA靶标功效预测的比较
为了评估DeepCRISPR在sgRNA靶标功效预测中的能力,我们首先为人类收集了完整的sgRNA靶标敲除功效基准数据,包括四种不同的细胞类型:hct116 [36],hek293t [2],hela [36] 和hl60 [37]。注意,Haeussler等人也使用了此类数据集为基准研究。整个数据集包含约15,000个sgRNA,这些经实验验证的有效敲除功效来自1071个基因。在我们的研究中,我们以分类模式或回归模式制定了DeepCRISPR,以进行全面而严格的比较。对于分类模型,已知的敲除功效以二进制方式标记(请参见“目标数据源”部分)。对于回归模型,以数字方式集成并标记了已知的敲除效率(请参见“目标数据源”部分)。然后,精心设计了八种不同的测试方案,以使用先进的工具对DeepCRISPR进行全面客观的比较。通过这种比较,我们提供了坚实的证据:
- 深度学习模型(无监督的预训练)优于浅层学习模型; 2.无监督的预训练策略可提高模型性能;
- 数据扩充进一步提高了模型性能和模型鲁棒性;
- DeepCRISPR在新细胞类型中普遍很好地用于sgRNA靶标敲除功效预测;
- DeepCRISPR避免了人工进行sgRNA设计的人工工程,从而有效地学习了高级特征表示,这与经过重新训练的sgRNA设计人员进行的apple-to-apple的比较表明(基于梯度升压的分类或回归模型) )具有相同的训练数据,但具有不同的功能;
- DeepCRISPR具有强大的分类和回归模型性能。
2.2.1 测试方案1-分类架构 classification schema
在该测试中,对于来自四种细胞类型的具有已知敲除效率的约15,000个sgRNA,每种数据类型的20%数据均通过数据标签进行了分层,并用作独立的测试集。来自每种细胞类型的其余80%数据组合在一起,以在交叉验证过程中进行模型训练和参数调整。在无监督的预训练和数据增强的情况下,基于深CNN的分类模型(表示为“ CNN”)分别针对四种细胞系的独立测试数据进行了训练和测试,并与八种状态进行了比较经过人类细胞系数据培训的先进工具,包括sgRNA Designer [2],SSC [5],CHOP-CHOP [10],CRISPR MultiTargeter [38],E-CRISP [9],sgRNA Scorer [12], Cas-Designer [39]和WU-CRISPR [40](图2a,b)。这些工具涵盖了通过学习模型或基于假设的评分功能为人类sgRNA功效预测设计的所有可用方法(有关当前工具的全面列表以及我们选择这些工具进行比较的原因,请参阅附加文件1)。使用来自接收器工作特性(ROC)曲线(AUC)[41]下面积的值评估比较。比较结果表明,平均而言,DeepCRISPR的总体ROC-AUC为0.796,优于所有八种方法,与sgRNA Designer [2]相比,最大提高了〜113%(以0.5 ROC-AUC为基线),是下一个性能最高的工具(图2a,b,附加文件2)。


2.2.2 测试方案2-分类架构
在该测试中,我们进一步对约6.8亿未标记的sgRNA(表示为“ pt CNN”)进行了无监督的预训练。 使用了与测试场景1相同的训练和测试数据。去除了训练和测试数据之间重叠的sgRNA。 预先训练的CNN的整体ROC-AUC达到0.836,比sgRNA设计器提高了142%(以0.5 ROC-AUC为基线;图2a,b;附加文件2)。
2.2.3 测试方案3-分类架构
我们进一步使用基于预训练的CNN和数据增强功能(称为“ pt + aug CNN”)构建了最终的DeepCRISPR模型。 扩增了训练数据,而测试数据与测试方案1和2相同。删除了训练和测试数据之间的重叠sgRNA。 对于这种情况,DeepCRISPR的总体ROC-AUC为0.857,比sgRNA设计器提高了157%(以0.5 ROC-AUC为基线;图2a,b;附加文件2)。 可以看出,与测试场景2相比,性能的提高相对较小,而我们发现训练过程中的损失函数收敛很快,并且与测试场景2相比变得非常健壮(图2c)。 这表明增加标签数据量可以帮助使模型更健壮并在训练过程中快速收敛。
2.2.4 测试方案4-分类架构
在这种情况下,我们进一步测试了DeepCRISPR在新细胞类型中的泛化能力。对于来自四种细胞类型的具有已知敲除效率的约15,000个sgRNA,每种数据类型的20%数据通过数据标签进行分层,并用作独立的测试集。来自不同细胞类型的其余80%的数据被作为训练数据进行了扩充,与测试场景3相同。然后,我们的模型以四重“留出一种细胞类型”的方式进行了训练,每次使用合并的训练数据从三种像元类型中进行测试,而将一种像元类型留在独立的数据集中进行测试。去除了训练和测试数据之间重叠的sgRNA。该测试方案研究了DeepCRISPR在新细胞类型上的泛化能力(图2d,附加文件2)。对于这种情况,DeepCRISPR在四种细胞类型上的性能平均ROC-AUC为0.722,优于第二好的方法sgRNA设计人员。可以看出,对于hct116和hela细胞类型,Deep- CRISPR的性能非常好。对于hek293t细胞类型,所有测试工具(包括DeepCRISPR)的性能均较差,这主要是由于这种细胞类型包含了大多数样品。因此,没有这种细胞类型数据的训练模型在训练数据不足的情况下效率低下。此外,为了研究特定于细胞类型的特征(即特定于细胞的表观遗传学特征)是否真正增加了DeepCRISPR的性能,我们重新训练了没有表观遗传学特征的DeepCRISPR(即,图2d)中仅Seq的Deep CRISPR模型用于性能比较)。可以看到在这种情况下,仅Seq的DeepCRISPR模型的性能与原始模型相比略有下降,这表明(1)细胞特异性表观遗传学特性确实增加了DeepCRISPR的性能,并且(2 HEK293T细胞类型),增加细胞特异性表观遗传学特征对预测性能的贡献似乎少于增加训练数据量的贡献。 DeepCRISPR在HL60细胞类型中表现中等。由于大多数其他工具(包括sgRNA设计工具,SSC等)都是根据HL60数据进行训练的,因此在这种特定细胞类型上,它们的性能通常优于DeepCRISPR。总而言之,我们得出结论,对于sgRNA靶标敲除功效预测,Deep-CRISPR在新细胞类型中通常表现良好。
2.2.5 测试方案5-分类架构
在此测试中,我们提供了DeepCRISPR与sgRNA Designer的更严格,更可靠的对比,它是我们先前测试中的次佳工具。 首先,我们为DeepCRISPR和sgRNA设计人员严格保留了相同的比较环境,并使用了相同的培训和测试数据。对于这种情况,我们使用与测试场景3中使用的Deep-CRISPR相同的增强标记数据集对 sgRNA Designer( https://github.com/MicrosoftResearch/Azimuth,一种基于梯度增强分类的浅层模型)进行了重新训练,并保留了测试数据相同。然后执行以下两个不同的特征表示:(1)我们用一键热特征表示(称为“经过训练的具有低级特征的sgRNA设计器”)编码sgRNA。该模型的总体ROC-AUC为0.751(图2a,b,附加文件2); (2)我们用sgRNA设计者采用的原始手动工程化特征(称为“经过培训的具有手动特征的sgRNA设计者”)对sgRNA进行了编码。该模型的总体ROC-AUC为0.778(图2a,b,附加文件2)。与这两种不同的特征表示相比,表明低级特征编码不适合浅层模型。因此,经过训练的sgRNA设计人员通过基于手动域的特征工程和特征编码获得了更好的性能。尽管如此,这些结果进一步表明,深度学习模型可以有效地从低级特征中学习高级特征表示,并且可以通过避免用于sgRNA设计的人工特征工程而与浅层模型竞争。 其次,我们还使用单热点特征表示法对DeepCRISPR和经过训练的sgRNA设计者进行了一种细胞类型的比较。该测试是在与测试场景4相同的训练和测试数据上进行的(图2d,附加文件2)。可以看出,平均而言,DeepCRISPR仍然比训练有素的sgRNA设计者表现更好,表明与其他方法相比,其在目标方面的预测优势。
2.2.6 测试方案6-回归模式 regression schema
在该测试中,我们以原始sgRNA基因敲除效率的回归模式对DeepCRISPR进行了培训。 如“目标数据源”部分所述,以优雅的方式集成了来自不同实验的数据。 使用Spearman相关性对性能进行评估,该相关性适用于以前的研究[42]。 整个比较以与测试场景3和5中类似的方式执行,不同之处在于以回归方式训练了模型。 同样,使用相同的训练和测试数据以回归方式对sgRNA设计者进行了再训练。 可以看出,在这种情况下,DeepCRISPR仍然优于Spearman相关性评估的其他方法(图2e,附加文件2)。
2.2.7 测试方案7-回归模式
我们进一步测试了基于回归的DeepCRISPR,以一种单细胞类型的方式研究其在新细胞类型中的泛化能力,类似于测试场景4。在这种情况下,DeepCRISPR的性能与分类方案中的相似,并且表现不佳。 由Spearman相关性评估的其他方法(图2f,附加文件2)。
2.2.8 测试方案8-独立数据集上的回归模式
由于之前的所有测试(方案1–7)都是通过分离数据进行训练和测试而对四种细胞类型(hct116,hek293t,hela和hl60)进行的,因此在本例中,我们应用了另一个数据集-完全独立于我们之前的测试,以研究DeepCRISPR的目标预测性能。最近通过利用荧光报告基因敲除测定法在选定的内源基因座进行sgRNA敲除功效测量的验证,报道了该数据集,共包含425个HEL细胞sgRNA。细胞类型和数据分布都与我们以前的测试不同,并且sgRNA与以前的数据集没有重叠。因此,它可以作为研究DeepCRISPR泛化能力的理想独立测试数据集。在此测试中,由于ENCODE中没有所测试的HEL细胞类型的表观遗传特征,因此我们在原始的四个细胞类型数据集上仅对具有序列级特征的DeepCRISPR进行了重新训练。重新训练的DeepCRISPR模型已在此HEL细胞数据上进行了测试,并与sgRNA设计器,SSC,sgRNA计分器和CRISPR进行了比较。令人惊讶的是,DeepCRISPR不仅明显胜过了当前最先进的靶标预测工具sgRNA designer,,在Spearmen相关性方面得到了近两倍的改进,而且也胜过了专门为此设计的CRISPRator HEL细胞数据集[43](图2g,附加文件3)。这项独立测试进一步表明,即使没有细胞类型特异性特征的贡献,DeepCRISPR对看不见的数据也具有良好的泛化能力。
总之,对于分类模型和回归模型,通过ROC-AUC和Spearman相关性进行测量,DeepCRISPR的性能通常优于针对目标预测的替代方法。而且,它具有良好的单元类型泛化能力。此外,可以看出训练数据的数量会影响模型的性能,并且可以通过使用大量训练数据来增强深度学习模型的潜力。
2.3 评估DeepCRISPR用于全基因组sgRNA脱靶谱预测
接下来,我们评估了DeepCRISPR预测脱靶位点的能力。为此,我们整理了由GUIDE-seq,Digenome-seq,BLESS,HTGTS和IDLV检测到的人类sgRNA全基因组脱靶谱数据。这些数据包括来自两种不同细胞类型的30种sgRNA:HEK 293细胞系及其衍生物(18 sgRNA)[6、22、24–27]和K562 t(12 sgRNA)[44],约有160,000个基因座最多有六个核苷酸错配(请参见“脱靶数据源”部分)。我们还通过分类和回归方案制定了DeepCRISPR,以进行全面而严格的比较。对于分类模型,将脱靶站点标记为“ 1”,其他标记为“ 0”(请参见“脱靶数据源”部分)。对于回归模型,脱靶位点标记有靶向效率,该靶向效率是由不同资产检测到的插入缺失频率测量的(请参见“脱靶数据源”部分)。然后针对脱靶轮廓预测评估设计了三种不同的测试方案。
2.3.1 测试场景1
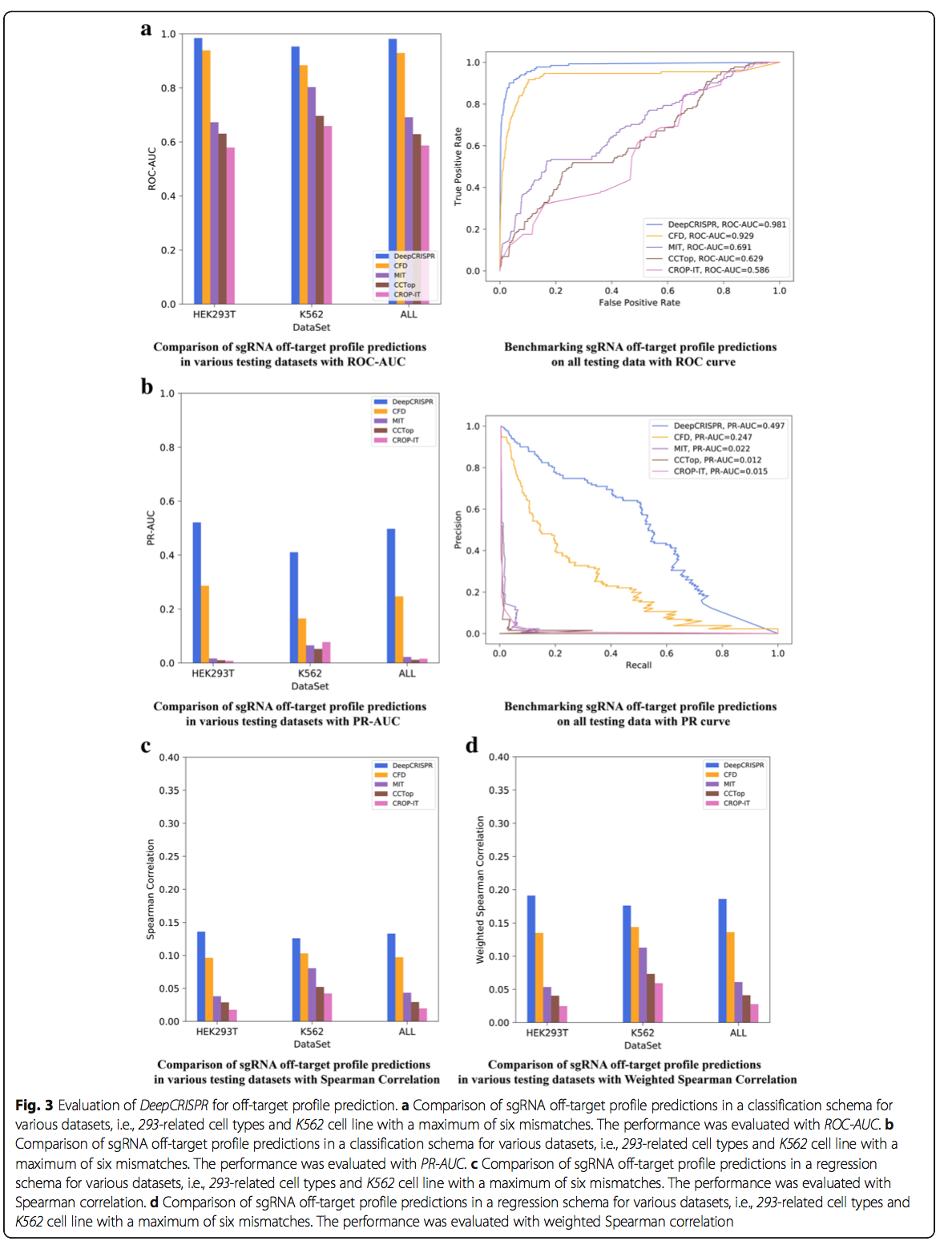
我们保留了每种细胞类型20%的数据作为独立测试集。其余80%的数据合并在一起以训练我们的模型并在交叉验证过程中调整参数。由于整个数据集在大约700个真正的脱靶站点之间高度失衡,因此在训练过程中采用了有效的自举采样算法,以减轻数据不平衡(请参阅“将自举集成到深度神经网络的批量训练中以解决数据不平衡问题”部分)。结果,我们在两个细胞系中的每一个的独立数据集上测试了我们最终训练的脱靶部位预测模型,并与四种最新的脱靶部位预测工具CFD得分[ 2],MIT评分[6],CROP-IT [45]和CCTop [46]。这些工具是根据各种经验定义的脱靶得分设计的,用于人类sgRNA脱靶位点预测。由于整个数据集是不平衡的,因此使用分类模型的ROC和精确召回曲线的AUC值以及回归模型的Spearman相关性和加权Spearman相关性[42]来评估比较。对于最多六个核苷酸错配,测试结果表明,DeepCRISPR在两种细胞类型中均胜过所有四种方法(图3a-c,附加文件2)。总体而言,DeepCRISPR的ROC-AUC为0.981(图3a),PR-AUC为0.497(图3b),Spearman相关系数为0.133(图3c),加权Spearman相关系数为0.186(图3c),优于第二好的方法,即CFD评分[2](图3a-c)。
值得注意的是,与使用ROC-AUC评估的CFD分数相比,使用DeepCRISPR进行脱靶预测的改善幅度相对较小,因为CFD分数已经实现了高性能。但是,这种改进非常重要,因为接近零的脱靶是所有基于CRISPR的基因治疗的最终目标。另外,应该指出的是,包括DeepCRISPR在内的所有现有工具都倾向于通过在阳性样品上增加权重来避免丢失真正的脱靶切割位点。这对于基于CRISPR的基因治疗也很有意义,因为错过真正的脱靶位点的惩罚总是比在脱靶位点预测中诱导假阳性的惩罚更高。这就是为什么我们采用Listgarten等人提出的加权Spearman相关性的原因。 [42]解决了这样的体重不对称问题。根据插入缺失频率测量的相应敲除功效,将每个脱靶位点的权重设置成与其等级顺序成正比。然而,这种加权方案实际上是对误报的一种折衷。因此,仍然需要仅从未加权数据中减少误报,这非常具有挑战性。对于这种情况,可以看出DeepCRISPR与其他方法相比大大提高了PR-AUC值,这表明DeepCRISPR可以大大减少脱靶预测期间的假阳性。
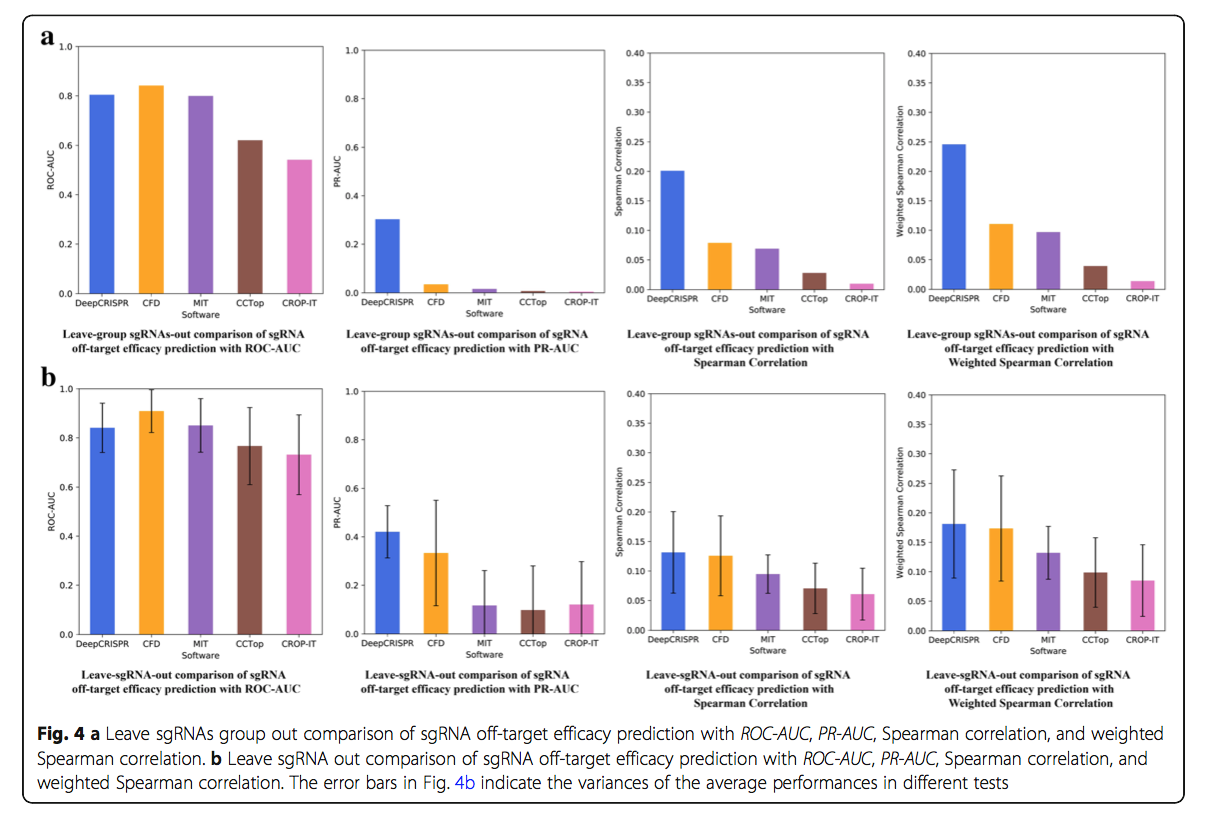
2.3.2 测试场景2
在这种情况下,对于来自两种不同细胞类型的所有30种sgRNA,我们执行了“将sgRNA分组出去”测试,这是脱靶谱检测的更具代表性的用例。这种测试随机地将一组sgRNA排除在外(在本例中为三个sgRNA)作为测试数据,从而提供了对一组看不见的sgRNA的预测性能的估计(图4a)。它可以确保一种sgRNA的脱靶指南完全位于测试组或训练集中。在这种情况下,对于分类模型和回归模型,DeepCRISPR的平均ROC-AUC为0.804,PR-AUC为0.303,Spearman相关性为0.201,加权Spearman相关性为0.246(图4a,附加文件2)。 DeepCRISPR的ROC-AUC可以与CFD分数相比较,而其他测量,特别是PR-AUC(0.303)则明显高于CFD分数(0.034),表明DeepCRISPR可以帮助降低脱靶预测中看不见的sgRNA的假阳性。
2.3.3 测试场景3
在这种情况下,对于所有30个sgRNA,我们进行了一次单次sgRNA外出测试的30倍,这是测试场景2中所示的离开sgRNA组外测试的一个极端案例(图4b)。对于分类模型和回归模型,DeepCRISPR的平均ROC-AUC为0.841,PR-AUC为0.421,Spearman相关性为0.132,加权Spearman相关性为0.181(图4b,附加文件2)。在这种情况下,DeepCRISPR的ROC-AUC与CFD评分的结果相当,而其他测量,尤其是PR-AUC(0.421),则高于CFD评分(0.333)。 总之,对于分类模型和回归模型,DeepCRISPR的性能通常都优于CFD评分,尤其是在降低失衡高度预测的假阳性方面,性能得到了改善。需要注意的一件事是,与回归模型相比,分类模型更适合于脱靶预测,因为在这种情况下,我们只关心区分脱靶位点,而不是预测其结合亲和力。此外,回归模型更敏感,因此需要更多数据来训练它。当前版本的DeepCRISPR仅在有限的样本上作为原型研究进行了培训。我们希望通过更多训练样本来增强DeepCRISPR,充分利用深层模型与浅层模型相比的优势。
2.4 在学习模式中自动进行特征识别
我们打算纯粹基于可用的训练数据和学习模型来自动化整个特征识别过程。 应当指出,基于浅层学习模型的功能识别和可视化已得到广泛解决,并且已经提出了许多工作来选择计算机模拟sgRNA设计的特征[1、2、5]。 但是,深度学习模型的特征识别和解释是不平凡的,值得探索。 在我们的研究中,我们提出了一种计算方法,可以基于训练有素的深度学习模型,通过优化[47],为有效的sgRNA设计推导特征显着图。 我们允许训练有素的深度神经网络模型来告诉我们,与无效sgRNA相比,有效sgRNA的外观如何(请参阅“通过推导特定类别的特征显着性图进行特征识别”一节)。
我们首先基于现有训练数据生成了sgRNA靶位预测的特征显着图,如图5a所示。我们获得了与以前的发现相符的特征显着性图 :
- 对于高效sgRNA,PAM NGG的可变核苷酸具有相同的偏好,其中偏爱胞嘧啶而胸腺嘧啶不利。这与现有的体外和体内研究一致[1,2]。
- 胸腺嘧啶在最靠近PAM的四个位置处不利,这与间隔区中的多个尿嘧啶导致sgRNA表达低的事实一致[1]。
- 位置18对胞嘧啶具有一致的偏好,胞嘧啶是CRISPR系统的DNA切割位点[5,37]。
- 它通常具有开放的染色质结构,如CTCF,DNase和H3K4me3的特征显着性图所示。
- 相对避免了DNA甲基化以实现高sgRNA功效,如RRBS分析所示。这与最近一项研究揭示了CRISPR脱靶的各种特征相一致[28]。
总之,在特定位置结合开放染色质结构的核苷酸偏好对于优化的sgRNA靶标设计是优选的。
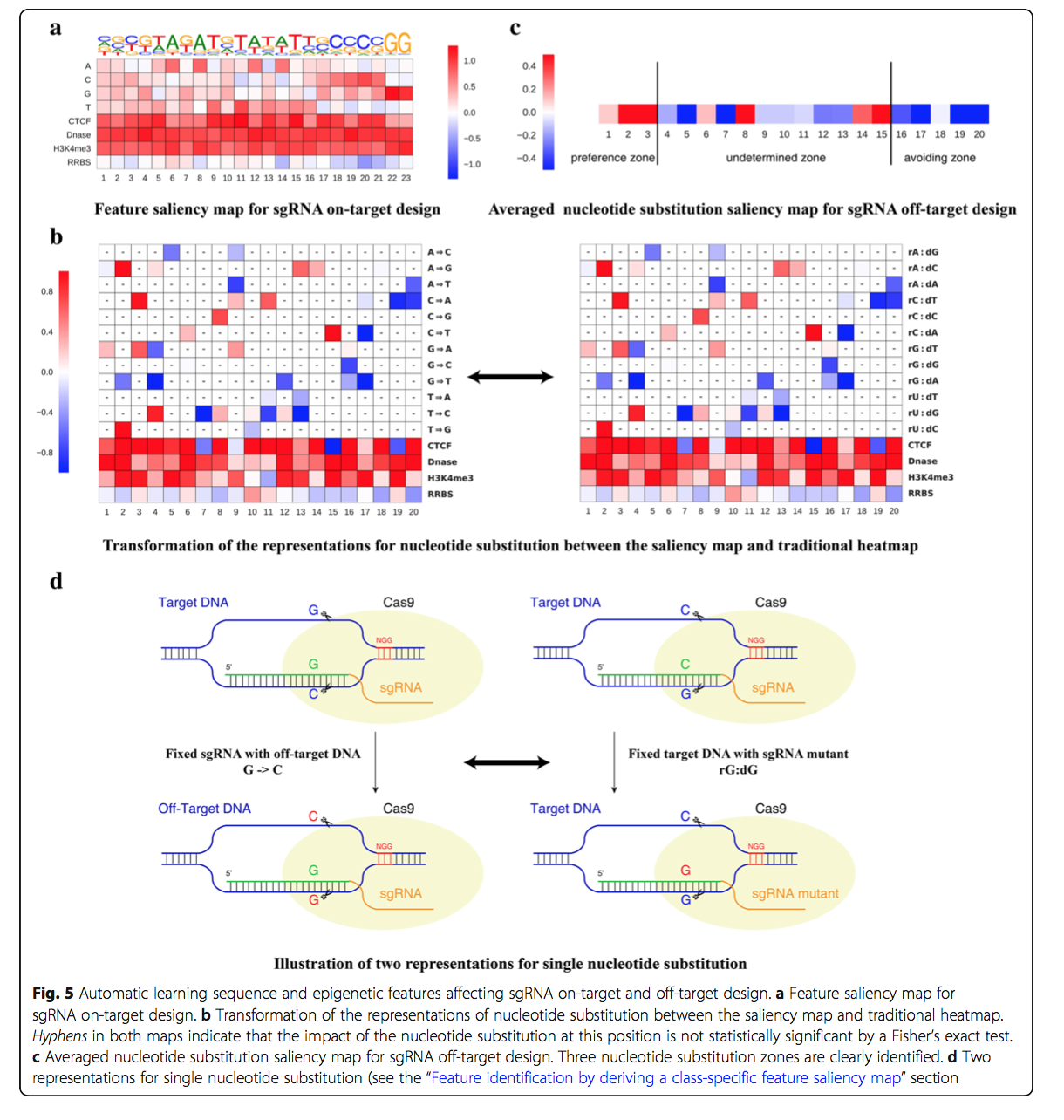
对于脱靶位点预测,通过使用Fisher统计检验过滤掉那些没有统计学显着性的点,从而给出了跨越20个位置的16种可能的核苷酸取代的详细特征显着图(图5b;参见“通过导出特定于类别的特征显着图”部分)。我们还生成了平均核苷酸取代率图,以表明它们对脱靶切割发生的影响(图5c;请参见“通过推导类特异性特征显着图进行特征鉴定”一节)。我们将该特征图划分为三个核苷酸取代区,即脱靶偏好区(位置1-3),未确定区(位置4-15)和脱靶避免区(位置16-20)。尽管此图是从有限的样本中获得的,但我们观察到,PAM附近发生的核苷酸突变易于以位置和核苷酸身份依赖性方式避免脱靶位点。这与先前的发现一致,即改变远离PAM的核苷酸通常对sgRNA的效力影响很小[2,6]。以前,两个不同的小组对人EMX1和CD33基因进行了广泛的体外测试,以产生不同的指导RNA,其中包含可能的单核苷酸取代,用于脱靶研究。他们的研究表明,SpCas9在PAM远端区域中的单碱基错配容忍程度大于PAM近端区域中的单碱基错配[2,6]。
具体来说,学习的特征图(附加文件4)给出的结果与以前的研究和新发现一致。 DeepCRISPR确定了对嘌呤的偏爱:-嘌呤错配以避免具有统计意义的脱靶位点,包括取代G-> C(对应于传统热图中的rG:dG,如先前报道[2])和取代G -> T(对应于传统热图中的rG:dA)在第16位的动态动力学研究表明,第16位的嘌呤和嘌呤错配会从根本上降低结合亲和力并降低裂解活性[ 48]。除了这些一致的发现外,我们的特征显着图还确定了在脱靶偏好区中有5个核苷酸取代更倾向于脱靶,而在脱靶避免区中有8种核苷酸取代避免了脱靶(图5b,c)。 ,包括位置16的两个核苷酸取代G-> C和G-> T [48]。这些基于大规模脱靶数据的核苷酸替代物的未来验证有望实现,并且将来随着脱靶数据的增加,所识别的因素将变得更加准确。 图5b的解释与以前的研究不同,在“通过导出类特定的特征显着图进行特征识别”部分对此进行了解释。
三、讨论和结论
在这里,我们介绍了DeepCRISPR,这是一个有效且可扩展的计算模型,用于同时预测CRISPR sgRNA的靶标敲除功效和全基因组靶标谱。 DeepCRISPR凭借可靠的评估指标,在各种人类数据集上超越了最先进的工具。 重要的是,我们的结果表明,利用全基因组范围内未标记的sgRNA序列以及深度学习模型有助于有效地学习sgRNA表示并提高预测性能。 此外,DeepCRISPR以数据驱动的方式自动进行sgRNA设计的特征识别,从而简化了解释,并优化了CRISPR靶向和脱靶设计。
参考资料
- 2018.Genome Biology. DeepCRISPR: optimized CRISPR guide RNA design by deep learning
