【3】数据分析-9-biopython实践
这两天处理数据的时候,需要解析Blast的结果。经管之前写过解析blast的xml结果的程序,用起来还是不够优雅。刚好,借这次机会来接触一下biopython。
安装
pip install biopython
Version 1.76 is the last release to support Python 2.7 and 3.5, all later releases require Python 3.6 or greater:
pip install biopython==1.76
一、简介
biopython是基于python语言来帮助生物信息学工作这解决问题的工具。它可以做:
- 将生物信息学文件解析为Python可用的数据结构,包含以下支持的格式:
- Blast输出结果 – standalone和在线Blast
- Clustalw
- FASTA
- GenBank
- PubMed和Medline
- ExPASy文件, 如Enzyme和Prosite
- SCOP, 包括‘dom’和‘lin’文件
- UniGene
- SwissProt
- 被支持格式的文件可以通过记录来重复或者通过字典界面来索引。
- 处理常见的生物信息学在线数据库的代码:
- NCBI – Blast, Entrez和PubMed服务
- ExPASy – Swiss-Prot和Prosite条目, 包括Prosite搜索
- 常见生物信息学程序的接口,例如:
- NCBI的Standalone
- Clustalw比对
- EMBOSS命令行工具
- 一个能处理序列、ID和序列特征的标准序列类。
- 对序列实现常规操作的工具,如翻译,转录和权重计算。
- 利用k最近邻接、Bayes或SVM对数据进行分类的代码。
- 处理比对的代码,包括创建和处理替换矩阵的标准方法。
- 分发并行任务到不同进程的代码。
- 实现序列的基本操作,翻译以及BLAST等功能的GUI程序。
- 使用这些模块的详细文档和帮助,包括此文件,在线的wiki文档,网站和邮件列表。
- 整合BioSQL,一个也被BioPerl和BioJava支持的数据库架构。 感觉好厉害的样子,还是从解析xml开始学起来吧。
biopython下载地址:http://biopython.org/wiki/Download
biopython官网说明:http://biopython-cn.readthedocs.io/zh_CN/latest/index.html
残基的识别
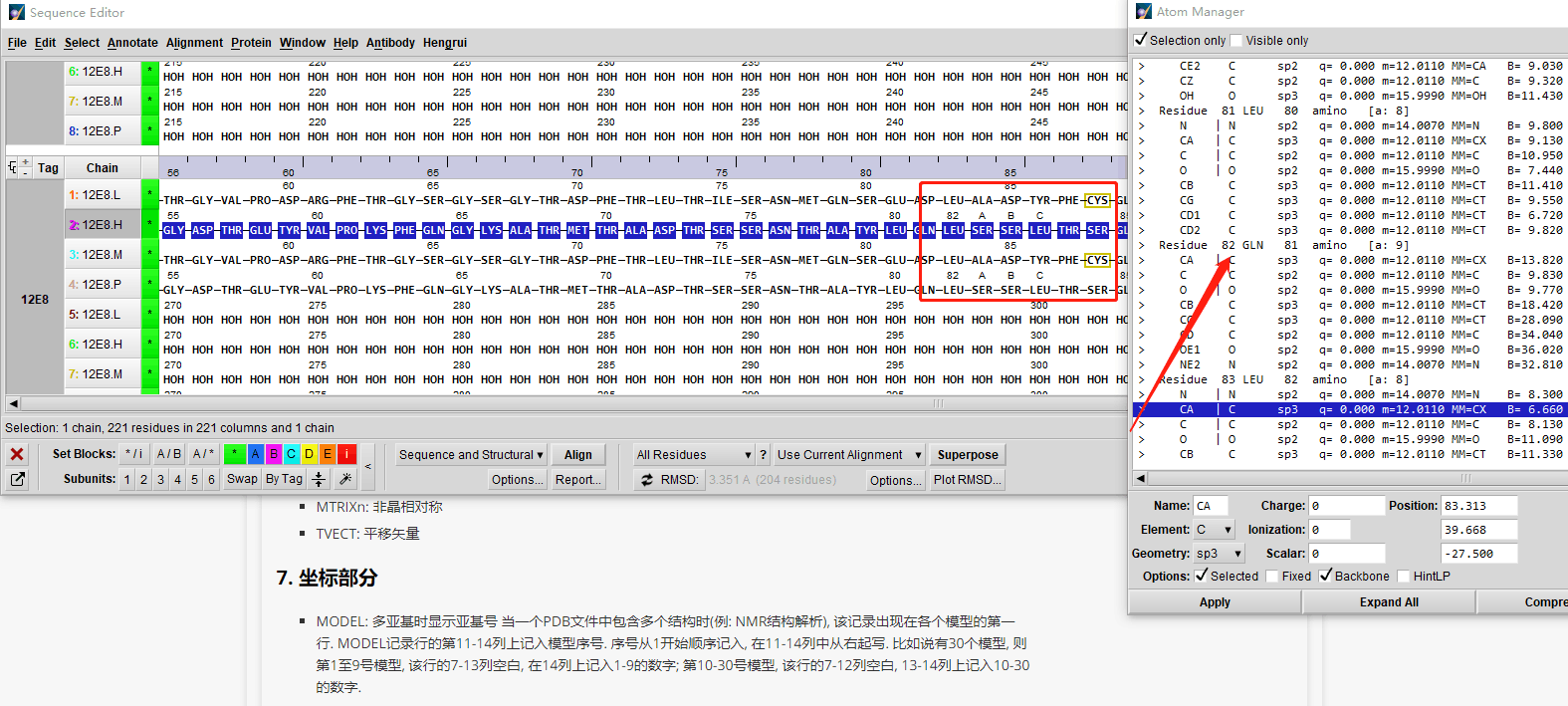
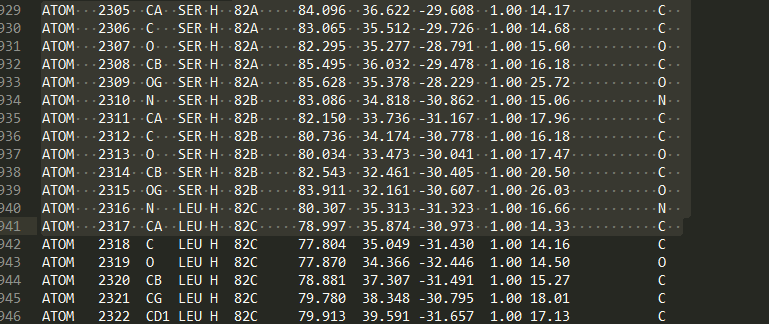
一个残基id是一个三元组:
- 异质域 (hetfield),即:
- ‘W’ 代表水分子
- ‘H_’ 后面紧跟残基名称,代表其它异质残基(例如 ‘H_GLC’ 表示一个葡萄糖分子)
- 空值表示标准的氨基酸和核酸 (关于异质残基的一个很普遍的问题是同一条链中的若干异质和非异质残基有同样的序列标识符(和插入码)。因此,要为每个异质残基生成唯一的id,水分子和其他异质残基应该以不同的方式来对待。)
- 序列标识符 (resseq),一个描述该残基在链上的位置的整数(如100);
- 插入码 (icode),一个字符串,如“A”。插入码有时用来保存某种特定的、想要的残基编号体制。一个Ser 80的插入突变(比如在Thr 80和Asn 81残基间插入)可能具有如下序列标识符和插入码:Thr 80 A, Ser 80 B, Asn 81。这样一来,残基编号体制保持与野生型结构一致。
因此,上述的葡萄酸残基id就是 (’H_GLC’, 100, ’A’) 。如果异质标签和插入码为空,那么可以只使用序列标识符:
# Full id
>>> residue=chain[(' ', 100, ' ')]
# Shortcut id
>>> residue=chain[100]
#full_id = residue.get_full_id()
("1abc", 0, "A", ("", 10, "A"))
#full_id = residue.get_id()
10
异质标签的起因是许许多多的PDB文件使用相同的序列标识符表示一个氨基酸和一个异质残基或一个水分子,这会产生一个很明显的问题,如果不使用异质标签的话。
毫不奇怪,一个残基对象存储着一个子原子集,它还包含一个表示残基名称的字符串(如 “ASN”)和残基的片段标识符(这对X-PLOR的用户来说很熟悉,但是在SMCRA数据结构的构建中没用到)。
让我们来看一些例子。插入码为空的Asn 10具有残基id (’ ’, 10, ’ ’) ;Water 10,残基id (’W’, 10, ’ ’);一个序列标识符为10的葡萄糖分子(名称为GLC的异质残基),残基id为 (’H_GLC’, 10, ’ ’) 。在这种情况下,三个残基(具有相同插入码和序列标识符)可以位于同一条链上,因为它们的残基id是不同的。
大多数情况下,hetflag和插入码均为空,如 (’ ’, 10, ’ ’) 。在这些情况下,序列标识符可以用作完整id的快捷方式:
# use full id
>>> res10 = chain[(' ', 10, ' ')]
# use shortcut
>>> res10 = chain[10]
一个链对象中每个残基对象都应该具有唯一的id。但是对含紊乱原子的残基,要以一种特殊的方式来处理,详见 11.3.3 。
一个残基对象还有大量其它方法:
>>> residue.get_resname() # returns the residue name, e.g. "ASN"
>>> residue.is_disordered() # returns 1 if the residue has disordered atoms
>>> residue.get_segid() # returns the SEGID, e.g. "CHN1"
>>> residue.has_id(name) # test if a residue has a certain atom
你可以用 is_aa(residue) 来检验一个残基对象是否为氨基酸。
获取序列中的残基:
one_residue_zz = one_residue.get_resname()
one_residue_simple = AA3_TO_AA1[one_residue_zz]
_aa_index = [('ALA','A'),
('CYS','C'),
('ASP','D'),
('GLU','E'),
('PHE','F'),
('GLY','G'),
('HIS','H'),
('HSE','H'),
('HSD','H'),
('ILE','I'),
('LYS','K'),
('LEU','L'),
('MET','M'),
('MSE','M'),
('ASN','N'),
('PRO','P'),
('GLN','Q'),
('ARG','R'),
('SER','S'),
('THR','T'),
('VAL','V'),
('TRP','W'),
('TYR','Y'),
('NRG','X')
]
二、实战
2.1.解析xml文件
from Bio.Blast import NCBIXML
def test_bio_python():
result_file = "tblastn_100000_2.xml"
output_file = "tblastn_100000_gene.tsv"
# with open(output_file,"w") as data1:
data2 = open(output_file, "w")
data2.write("seq\tlength\tgen\tmap_len\tmismatch\n")
result_handle = open(result_file)
blast_records = NCBIXML.parse(result_handle)
blast_records = list(blast_records)
for blast_record in blast_records:
# blast_record为一条序列的比对结果
# for description in blast_record.descriptions:
# print description.title
# print description.score
# print description.e
# print description.num_alignments
# break
# print blast_record.
# print blast_record.description.title
# print dir(blast_record) 可以看到每条blast_record的基本属性
query_id = blast_record.query # query的名字
query_length = blast_record.query_length
# print blast_record.reference
# print blast_record.query_letters
# print blast_record.query_id
for aligment in blast_record.alignments:
#aligment为一条序列比对到一个基因,其中一个基因有可能被多次Hits
for hsp in aligment.hsps:
# print hsp.__getattr__
# print aligment.title #被Hit的序列的名字,相当于xml中的
#<Hit_def>Homo sapiens ER membrane protein complex subunit 1 (EMC1), transcript variant 2, mRNA</Hit_def>
#获得()中的基因名字
if "(" in aligment.title:
if aligment.title.count("(") == 1:
gene = aligment.title.split("(")[1].split(")")[0]
elif aligment.title.count("(") == 2:
gene = aligment.title.split(")")[1].split("(")[1]
else:
gene = aligment.title
# print aligment.length #比对序列的长度
# print hsp.score
# print hsp.bits
# print hsp.expect
# print hsp.num_alignments
# print hsp.identities
identity_len = hsp.identities #比对到的碱基个数,相当于<Hsp_identity>4</Hsp_identity>
# print hsp.positives #相当于<Hsp_positive>6</Hsp_positive>
# print hsp.gaps #相当于<Hsp_gaps>0</Hsp_gaps>
# print hsp.strand
# print hsp.frame
# print hsp.query #相当于<Hsp_qseq>LLVDDQ</Hsp_qseq>
# print hsp.query_start
# print hsp.match #相当于<Hsp_midline>LL+DD+</Hsp_midline>
# print hsp.sbjct #相当于<Hsp_hseq>LLIDDE</Hsp_hseq>
# print hsp.sbjct_start
gaps = hsp.gaps
identity_len = hsp.identities
align_len = len(query_seq) - int(gaps)
identity_1 = int(identity_len) / int(align_len) * 100 #比对上的序列的相似性
identity_2 = int(identity_len) / int(query_length) * 100 #全长比对的百分比
mismatch = int(query_length) - int(identity_len)
result = [query_id, str(query_length), gene, str(identity_len),
str(mismatch)]
data2.write("%s\n" % "\t".join(result))
# break
data2.close()
后面用到,再来接着补充吧
2.蛋白pdb文件的分析
下载蛋白序列文件
from Bio.PDB.PDBList import PDBList
def download_pdb(pdb_id, download_dir='pdb'):
pdbl = PDBList()
pdf_name = os.path.join(download_dir, 'pdb%s.ent' % pdb_id.lower())
if not os.path.exists(pdf_name):
pdbl.retrieve_pdb_file(pdb_id, pdir=download_dir, file_format='pdb')
return pdf_name
根据指定位置提取残基的向量
from Bio.PDB.PDBParser import PDBParser
def get_vector(pdb_id, model, chain, chain_s, chain_e, download_dir='pdb'):
structure_info = []
parser = PDBParser()
pdf_name = download_pdb(pdb_id, download_dir)
chain_info = parser.get_structure("test", pdf_name)[model][chain]
#for ii in range(chain_s, chain_e + 1):
# 注意这里了,这个地方我一直报错,原因在于hetatm这一行读为residue以后;residue的key变成了('H_MSE', ii, ' '),而不再是(' ', ii, ' '),所以,就不能直接用chain_info[ii]来提取了,当然如果这个残基不是MSE,就需哟啊灵当别论了
# try :
# atom = chain_info[ii]['CA']
# except:
# atom = chain_info[('H_MSE', ii, ' ')]['CA']
# atom = chain_info[ii]['CA']
# vector_info = atom.get_vector()
#structure_info.append(list(vector_info))
locs_range = range(chain_s, chain_e + 1)
for one_residue in chain_info:
residue_id = one_residue.get_id()[1]
if residue_id in locs_range :
atom = one_residue['CA']
vector_info = atom.get_vector()
structure_info.append(list(vector_info))
return structure_info
3.修改指定参加的所有原子的向量
这里面涉及到怎么通过biopython写PDB文件,怎么提取指定的atom,并如何修改atom,短短几行代码,做了如此多的事情,精妙呀
from Bio.PDB.PDBParser import PDBParser
from Bio.PDB.PDBList import PDBList
from Bio.PDB.PDBIO import PDBIO,Select
from Bio.PDB import extract
class SelectAtom(Select):
def __init__(self,chain_s,chain_e,new_vector):
self.chain_s = chain_s
self.chain_e = chain_e
self.new_vector = new_vector
def accept_residue(self,residue):
id_name = residue.get_id()[1]
if id_name < self.chain_s or id_name > self.chain_e:
return False
else:
for atom in residue :
atom.set_coord(self.new_vector)
return True
def get_atom(pdb_id, model, chain, chain_s, chain_e, download_dir='pdb'):
parser = PDBParser()
pdf_name = download_pdb(pdb_id, download_dir)
chain_info = parser.get_structure("test", pdf_name)[model][chain]
new_list = [2,2,2]
io = PDBIO()
io.set_structure(chain_info)
io.save('out.pdb',SelectAtom(chain_s,chain_e,new_list))
2.3 读fasta序列
from Bio import SeqIO
fastas = SeqIO.parse(fasta_file, 'fasta')
for fasta in fastas:
print fasta.id # 如果序列名有空格,只识别第一个空格前的内容
print fasta.description # 整个序列名
print fasta.seq
生成的序列固定长度
while len(sequence) > 0:
# print(sequence)
new_seq = sequence[:width]
data1.write('%s\n' % new_seq)
sequence = sequence[width:]
2.4 核酸序列转氨基酸序列
from Bio.Seq import Seq
seq = Seq(test_seq)
print seq.translate()
2.4.2 从PDB中提取序列
根据结构来提取序列,如果结构断裂,则序列会断裂
>>> from Bio.PDB.PDBParser import PDBParser
>>> from Bio.PDB.Polypeptide import PPBuilder
>>> structure = PDBParser().get_structure('1A8O', 'PDB/1A8O.pdb')
>>> ppb=PPBuilder() # 根据CN距离,如果根据CA距离,则CaPPBuilder
>>> for pp in ppb.build_peptides(structure):
... print(pp.get_sequence())
DIRQGPKEPFRDYVDRFYKTLRAEQASQEVKNW
TETLLVQNANPDCKTILKALGPGATLEE
TACQG
如下图所示的非标氨基酸Tys,则需要加入aa_only=False
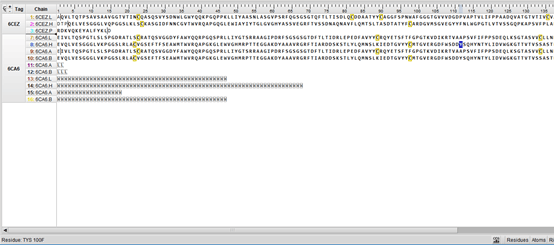
>>> for pp in ppb.build_peptides(structure, aa_only=False):
... print(pp.get_sequence())
... print("%s %s" % (pp.get_sequence()[0], pp[0].get_resname()))
... print("%s %s" % (pp.get_sequence()[-7], pp[-7].get_resname()))
... print("%s %s" % (pp.get_sequence()[-6], pp[-6].get_resname()))
MDIRQGPKEPFRDYVDRFYKTLRAEQASQEVKNWMTETLLVQNANPDCKTILKALGPGATLEEMMTACQG
M MSE
M MSE
M MSE
2.5 PDB提取某些链,并修改链的名字
# 提取链,并修改名字
class SelectChain(Select):
def __init__(self,chain_hash):
self.chain_hash = chain_hash
def accept_chain(self,chain):
id_name = chain.get_id()
if id_name not in self.chain_hash:
return False
else:
chain.id = self.chain_hash[id_name] # 修改链的名字
return True
# 只提取链
class SelectChain_2(Select):
def __init__(self,chain_hash):
self.chain_hash = chain_hash
def accept_chain(self,chain):
id_name = chain.get_id()
if id_name not in self.chain_hash:
return False
else:
return True
# pdb_id: '4XGZ'
# chain_hash:{'A':'H','B':'L'}
def keep_chain(pdb_id,chain_hash,result_dir='./'):
pdf_name = download_pdb(pdb_id)
parser = PDBParser()
chain_info = parser.get_structure("test", pdf_name)
io = PDBIO()
io.set_structure(chain_info)
pdb_result = '%s/%s.pdb'% (result_dir,pdb_id)
pdb_result_2 = '%s/%s_2.pdb'% (result_dir,pdb_id)
try:
io.save(pdb_result,SelectChain(chain_hash))
except:
io.save(pdb_result_2, SelectChain_2(chain_hash))
parser_2 = PDBParser()
chain_info_2 = parser_2.get_structure("test", pdb_result_2)
io_2 = PDBIO()
io_2.set_structure(chain_info_2)
pdb_result = '%s/%s.pdb' % (result_dir, pdb_id)
io_2.save(pdb_result, SelectChain(chain_hash))
os.system('rm %s' % pdb_result_2)
注:先尝试提取链的时候,也修改链的名字,但是有的PDB修改后的名字已经存在其他链中,就会报错:
ValueError: Cannot change id from `A` to `h`. The id `h` is already used for a sibling of this entity.
所以,先提取,提取以后再修改
2.6 修改残基的ID
class ChangeAtomUid(Select):
"""
new_id_hash: eg. { "model-chain-id-label":{'id':100,'icode':'A' }) }
"""
def __init__(self,new_id_hash,accpet_chain='na'):
self.new_id_hash = new_id_hash
self.renames = {}
self.chains = accpet_chain
def accept_residue(self,residue):
full_id = residue.get_full_id() ## ("1abc", 0, "A", ("", 10, "A"))
if full_id[2] in self.chains or self.chains == 'na':
id_full = '%s-%s-%s-%s' % (full_id[1]+1,full_id[2],full_id[3][1],full_id[3][2].strip())
if id_full in self.renames:
id_1 = self.renames[id_full]
else:
id_1 = id_full
if id_1 in self.new_id_hash:
new_id = (full_id[3][0], self.new_id_hash[id_1]['id'], self.new_id_hash[id_1]['icode'])
tmp_id = ('',1000+self.new_id_hash[id_1]['id'],'%s' % self.new_id_hash[id_1]['icode'])
tmp_full_id = '%s-%s-%s-%s' % (full_id[1] + 1, full_id[2], 1000+self.new_id_hash[id_1]['id'], '%s' % self.new_id_hash[id_1]['icode'].strip())
try:
residue.id = new_id
except:
full_id_2 = residue.get_parent()[new_id].get_full_id() ## ("1abc", 0, "A", ("", 10, "A"))
id_new = '%s-%s-%s-%s' % (full_id_2[1] + 1, full_id_2[2], full_id_2[3][1], full_id_2[3][2].strip())
self.renames[tmp_full_id] = id_new
residue.get_parent()[new_id].id = tmp_id # 先转成tmp id,避免ID重复
residue.id = new_id
return True
else:
return False
2.7 二硫键的分析
- The sulfur atoms of Cys amino-acids often form “di-sulfide” bonds if they are close enough – less than 8 Å.
- Compare with PDB file contents: SSBOND
- Bio.PDB does not provide an easy way to access the SSBOND annotations
In a protein in the oxidative environment, a covalent bond is often formed between a sulfur atom of two Cys residues, when the sulfur atoms exist in a short distance. The bond is called disulfide bond or S-S bond in short. A disulfide bond plays an important role in protein folding and stability.
In proteins with the known three-dimensional structure, the distances of two bonded sulfur atoms is known to be distributed between 1.8 - 2.0Å with a highest frequency at about 2.0 Å and dihedral angles of C-S-S-C around 90º.
import Bio.PDB.PDBParser
import sys
# Use QUIET=True to avoid lots of warnings...
parser = Bio.PDB.PDBParser(QUIET=True)
structure = parser.get_structure("1KCW", "1KCW.pdb")
model = structure[0]
achain = model['A']
cysresidues = []
for residue in achain:
if residue.get_resname() == 'CYS':
cysresidues.append(residue)
for c1 in cysresidues:
c1index = c1.get_id()[1]
for c2 in cysresidues:
c2index = c2.get_id()[1]
if (c1['SG'] - c2['SG']) < 8.0:
print "possible di-sulfide bond:",
print "Cys",c1index,"-",
print "Cys",c2index,
print round(c1['SG'] - c2['SG'],2)
http://cib.cf.ocha.ac.jp/bitool/SSBOND/
2.7 寻找指定距离内的两个原子
from Bio.PDB import PDBParser,Selection,NeighborSearch
def get_all_neighbors(input_pdb,distance_cutoff=3.5):
structure = PDBParser().get_structure('test', input_pdb)
atom_list = Selection.unfold_entities(structure, 'A') # (A=atoms, R=residues, C=chains, M=models, S=structures).
ns = NeighborSearch(atom_list)
all_neighbors = ns.search_all(float(distance_cutoff), "R")
pair_neighbors = []
for residuepair in all_neighbors:
one_pair = []
for one_residue in residuepair:
one_pair.append('%s-%s-%s' % (
one_residue.get_full_id()[2], one_residue.get_full_id()[3][1], one_residue.get_full_id()[3][2].strip()))
pair_neighbors.append(one_pair)
return pair_neighbors
2.8 Find contact residues in a dimer – better version
import Bio.PDB.PDBParser
import sys
# Use QUIET=True to avoid lots of warnings...
parser = Bio.PDB.PDBParser(QUIET=True)
structure = parser.get_structure("1HPV","1HPV.pdb")
achain = structure[0]['A']
bchain = structure[0]['B']
bchainca = [ r['CA'] for r in bchain ]
neighbors = Bio.PDB.NeighborSearch(bchainca)
for res1 in achain:
r1ca = res1['CA']
r1ind = res1.get_id()[1]
r1sym = res1.get_resname()
for r2ca in neighbors.search(r1ca.get_coord(), 6.0):
res2 = r2ca.get_parent()
r2ind = res2.get_id()[1]
r2sym = res2.get_resname()
print "Residues",r1sym,r1ind,"in chain A",
print "and",r2sym,r2ind,"in chain B",
print "are close to each other:",round(r1ca-r2ca,2)
2.9 Superimpose two structures
import Bio.PDB
import Bio.PDB.PDBParser
import sys
# Use QUIET=True to avoid lots of warnings...
parser = Bio.PDB.PDBParser(QUIET=True)
structure1 = parser.get_structure("2WFJ","2WFJ.pdb")
structure2 = parser.get_structure("2GW2","2GW2a.pdb")
ppb=Bio.PDB.PPBuilder()
# Manually figure out how the query and subject peptides correspond...
# query has an extra residue at the front
# subject has two extra residues at the back
query = ppb.build_peptides(structure1)[0][1:]
target = ppb.build_peptides(structure2)[0][:-2]
query_atoms = [ r['CA'] for r in query ]
target_atoms = [ r['CA'] for r in target ]
superimposer = Bio.PDB.Superimposer()
superimposer.set_atoms(query_atoms, target_atoms)
print "Query and subject superimposed, RMS:", superimposer.rms
superimposer.apply(structure2.get_atoms())
# Write modified structures to one file
outfile=open("2GW2-modified.pdb", "w")
io=Bio.PDB.PDBIO()
io.set_structure(structure2)
io.save(outfile)
outfile.close()
2.10 Superimpose two chains
import Bio.PDB
parser = Bio.PDB.PDBParser(QUIET=1)
structure = parser.get_structure("1HPV","1HPV.pdb")
model = structure[0]
ppb=Bio.PDB.PPBuilder()
# Get the polypeptide chains
achain,bchain = ppb.build_peptides(model)
aatoms = [ r['CA'] for r in achain ]
batoms = [ r['CA'] for r in bchain ]
superimposer = Bio.PDB.Superimposer()
superimposer.set_atoms(aatoms, batoms)
print "Query and subject superimposed, RMS:", superimposer.rms
superimposer.apply(model['B'].get_atoms())
# Write structure to file
outfile=open("1HPV-modified.pdb", "w")
io=Bio.PDB.PDBIO()
io.set_structure(structure)
io.save(outfile)
outfile.close()
参考资料
