【6.1.2.3】bedtools
bedtools是一款针对BED, VCF, BAM各种处理的工具,强大的超乎想象。
一、Bedtools简介
BEDTools是可用于genomic features的比较,相关操作及进行注释的工具。而genomic features通常使用Browser Extensible Data (BED) 或者 General Feature Format (GFF)文件表示,用UCSC Genome Browser进行可视化比较。
安装 :http://bedtools.readthedocs.io/en/latest/content/installation.html
下载地址:https://code.google.com/p/bedtools/downloads/list
git://github.com/arq5x/bedtools.git
1. 解压缩
2.Cd到解压缩文件中
3.Make
4.如果遇到问题,联系aaronquinlan at gmail dot com
5.运行 make test
6. Copy the files in bin/ to ~/bin or if you have the privileges, to /usr/local/bin.
Make sure that the directory to which you copy the tools is in your $PATH (我在其他的地方运行了一下,居然可以用,不知道是不是之前就已经安装好了,这一步就没有做)
7. Use the tools.
$ wget https://github.com/arq5x/bedtools2/releases/download/v2.25.0/bedtools-2.25.0.tar.gz
$ tar -zxvf bedtools-2.25.0.tar.gz
$ cd bedtools2
$ make
二、常用命令
2.1.genocov
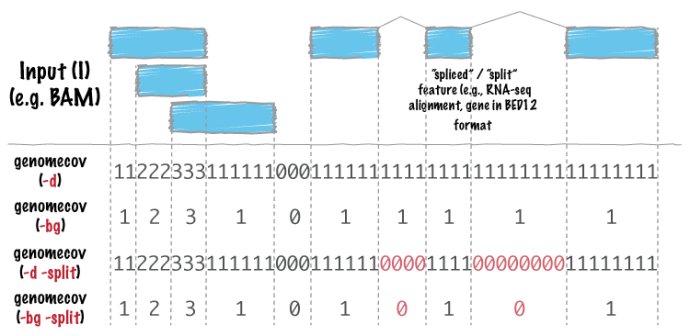
在终端输入 bedtools genomecov 你可以看到具体的说明,具体的我就不说了
Tool: bedtools genomecov (aka genomeCoverageBed)
Version: v2.17.0
Summary: Compute the coverage of a feature file among a genome.
Usage: bedtools genomecov [OPTIONS] -i -g
Options:
Notes:
(1) The genome file should tab delimited and structured as follows:
For example, Human (hg19):
chr1 249250621
chr2 243199373
...
chr18_gl000207_random 4262 #关键是这一步,我需要提取我的scaffold文件名和长度,在这一步理解的不够好,花了点时间啊
(2) The input BED (-i) file must be grouped by chromosome.
A simple "sort -k 1,1 > .sorted" will suffice.
(3) The input BAM (-ibam) file must be sorted by position.-ibam 输入文档必须是BAM格式,同时也要是排序后的BAM文件
A "samtools sort " should suffice.
Tips:
One can use the UCSC Genome Browser's MySQL database to extract
chromosome sizes. For example, H. sapiens:
mysql --user=genome --host=genome-mysql.cse.ucsc.edu -A -e \
"select chrom, size from hg19.chromInfo" > hg19.genome
达到目的的最后命令:
对scaffold.fa前处理
cd /sam/idba_ud/myproject/clean_reads/index
grep 'scaffold' scaffold.fa>scaffold.txt
cut -f1,2 -d " " scaffold.txt>scaffold.txt2
cat scaffold.txt2 |sed 's/>scaffold/scaffold/g'|sed 's/length_//g' >scaffold.txt3
获得coverage
bedtools genomecov -ibam eg2.sorted.bam -g scaffold.txt3 > coverage.txt
出来的结果:第一列是scaffold,第二列是起始位置,第三列是终止位置,第四列是总大小,第五列是覆盖度
在genomecov里面,第三列是总共覆盖的碱基数目
bedtools genomecov -ibam GWB0514095_sort.primerclipped.realign.recalibrate.bam -bg |grep 4119781
17 41197808 41197811 5802
17 41197811 41197812 5800
17 41197812 41197821 5802
因为得到的是真实的bed文件,实际上是17:41197809-41197811:5802
2.2 intersect
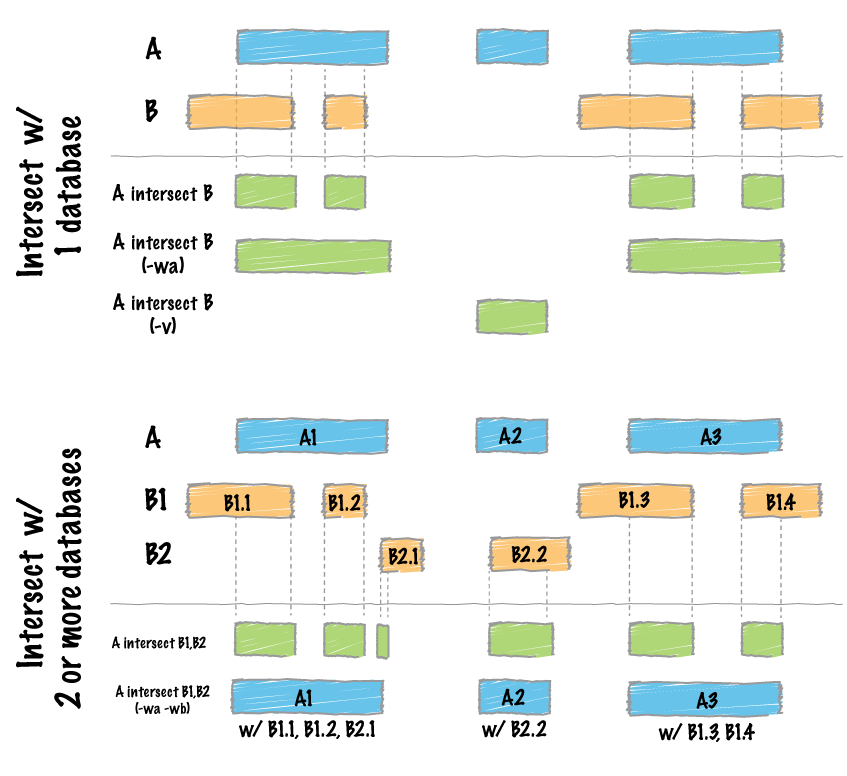
head BRCAWise_LP.exons.bed
17 41276033 41276113 BRCA1_2
17 41267742 41267796 BRCA1_3
17 41258472 41258550 BRCA1_4
17 41256884 41256973 BRCA1_5
17 41256138 41256278 BRCA1_6
17 41251791 41251897 BR 17 41249260 41249306 BRCA1_8
17 41247862 41247939 BRCA1_9
17 41243451 41246877 BRCA1_10
17 41242960 41243049 BRCA1_11
head XYC6640369.cut30.merged.reg
13 32890555 32890722
13 32893188 32893484
13 32899161 32899413
13 32900230 32900481
13 32900604 3290
13 32903515 32903669
13 32904990 32905201
13 32906351 32907673
13 32910325 32915349
13 32918580 8907
bedtools intersect -a BRCAWise_LP.exons.bed -b XYC6640369.cut30.merged.region -wao |head
17 41276033 41276113 BRCA1_2 17 41275967 41276121 80
17 41267742 41267796 BRCA1_3 17 41267681 41267884 54
17 41258472 41258550 BRCA1_4 17 41258405 41258611 78
17 41256884 41256973 BRCA1_5 17 41256823 41256991 89
17 41256 41256278 BRCA1_6 17 41256048 41256343 140
17 41251791 41251897 BRCA1_7 17 41251686 41251957 106
17 41249260 41249306 BRCA1_8 17 41249203 41249372 46
17 41247862 41247939 BRCA1_9 17 41247839 41247993 77
17 41243451 41246877 BRCA1_10 17 41243387 41246958 3426
17 41242960 41243049 BRCA1_11 17 41242918 41243083 89
bedtools intersect -f 0.95 -a NA14624-25-2_sort.bed -b test2.bed
-f 0.95 代表-a中至少有95%的被-b的overlap掉,才算overlap
bedtools intersect -f 0.95 -abam NA14624-25-2_sort.bam -b test2.bed >test11.bam
2.3 merge
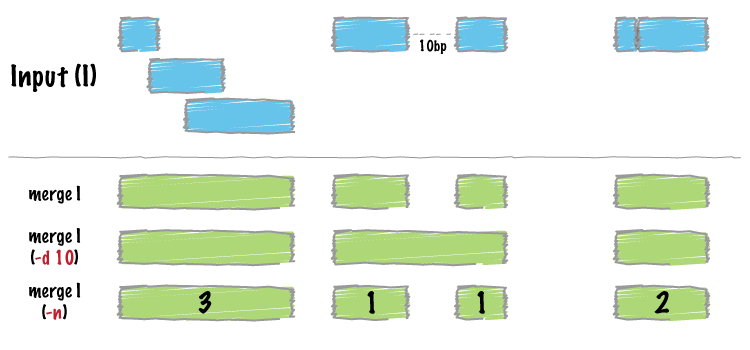
统计合并后的bed
bedtools merge -i unmapped_bed/GA-sort.unmapped.bed -c 4 -o count > count_bed/GA-sort.region.be
2.4 comple
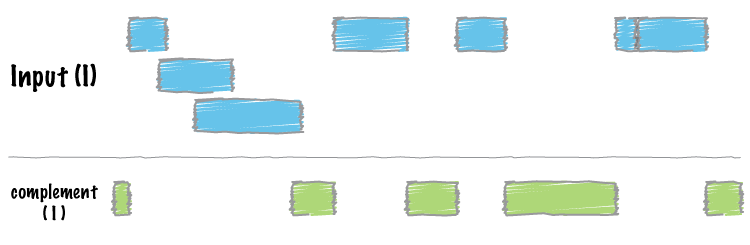
2.5 subtra
subtract 求非交集 –a –b 得到的是a没有被b覆
bedtools subtract -a BRCAWise_LP.exons.bed -b XYC6640369.cut30.merged.region |head
13 4693 32944694 BRCA2_19
扩增子区域跟q20重叠的区域,求覆盖
第二列是amplicon的起始位置,第三列是amplicon终止位置
amplicon被覆盖的碱基个数为最后一列
2.6 coverage
http://bedtools.readthedocs.io/en/latest/content/tools/coverage.h
$ cat A.bed
chr1 0 100
chr1 100 200
chr2 0 100
$ cat B
chr1 10 20chr1 20 30chr1 30 40
chr1 100 200
$ bedtools coverage -a A.bed -b B.bed
chr100 3 30 100 0.3000000
chr1 100 200 1 100 100 1.0000000
chr2 0 100 0 0 100 0.0000000
-s 计算正反链
$ cat A.bed
chr1 0 100 b1 1 +
chr1 100 200 b2 1 -
chr2 0 100 b3 1 +
$ cat B.bed
chr1 10 20 a1 1 -
chr1 20 30 a2 1 -
chr1 30 40 a3 1 -
chr1 100 200 a4 1 +
$ bedtools coverage -a A.bed -b B.bed
chr1 0 100 b1 1 + 3 30 100 0.3000000
chr1 100 200 b2 1 - 1 100 100 1.0000000
chr2 0 100 b3 1 + 0 0 100 0.0000000
$ bedtools coverage -a A.bed -b B.bed -s
chr1 0 100 b1 1 + 0 0 100 0.0000000
chr1 100 200 b2 1 - 0 0 100 0.0000000
chr2 0 100 b3 1 + 0 0 100 0.0000000
计算amplicon的depth
-
将bed中成对的read对应的amplicon start和end提出来,一对reads会有两行
awk ‘BEGIN{OFS ="\t"} {if($9<0){print $3,$8,$8-$9+1,$1} else if($9>0){print $3,$4,$4+$9-1,$1}}’ GWA5720199.sam >GWA5720199.amplicon.bed
-f 代表两者的匹配百分比 -r 代表两个文件互相匹配
-
提取amplicon对应的reads数
bedtools coverage -b GWA5720199.amplicon.bed -a brca.target.amplicon.bed -f 0.96 -r >test2.depth
test2.depth文件如下: chrom start end amplicon_name reads(单条的read) amplicon length 17 41197639 41197852 BRCA1_1 781 213 213 1.0000000 17 41197642 41197817 BRCA1_2 822 175 175 1.0000000 17 41197713 41197869 BRCA1_3 298 156 156 1.0000000 17 41199563 41199766 BRCA1_4 830 203 203 1.0000000
最新版bedtools v2.26.0才有-f 和-r参数
-f Minimum overlap required as a fraction of A. Default is 1E-9 (i.e. 1bp).
-r Require that the fraction of overlap be reciprocal for A and B. In other words, if -f is 0.90 and -r is used, this requires that B overlap at least 90% of A and that A also overlaps at least 90% of B.
2.7 maskfasta
bedtools maskfasta [OPTIONS] -fi <input FASTA> -bed <BED/GFF/VCF> -fo <output FASTA>
或者可以这样:
maskFastaFromBed [OPTIONS] -fi <input FASTA> -bed <BED/GFF/VCF> -fo <output FASTA>
# The input (-fi) and output (-fo) FASTA files must be different.
Option Description
-soft Soft-mask (that is, convert to lower-case bases) the FASTA sequence. By default, hard-masking (that is, conversion to Ns) is performed.
-mc Replace masking character. That is, instead of masking with Ns, use another character.
参考资料:http://bedtools.readthedocs.io/en/latest/content/tools/maskfasta.html
三、报错
3.1 安装过程中报错
centos编译时报错:error: bzlib.h: No such file or directory
解决方法:
1.首先查看你yum源中的bzip2包,命令为:
yum search bzip2
2.安装你yum源中的bzip2包,执行命令:
yum install bzip2-devel.x86_64
3.2 安装过程中报错
gcc -g -Wall -O2 -I. -c -o cram/cram_io.o cram/cram_io.c
cram/cram_io.c:61:18: fatal error: lzma.h: No such file or directory
#include <lzma.h>
^
compilation terminated.
make[1]: *** [cram/cram_io.o] Error 1
make[1]: Leaving directory `/data/software/bedtools2/src/utils/htslib'
make: *** [src/utils/htslib/libhts.a] Error 2
解决办法:
配置好yum源后安装xz-devel包,执行命令为:
yum install -y xz-devel
四、讨论
1.计算amplicon的深度(amplicon之间有overlap的情况,但这种方法比较耗内存,也比较慢,不是太建议)
os.system("{samtools} view {input_bam}>{sam_file}".format(**locals()))
cmd1 = """
awk 'BEGIN{OFS ="\t"} {if($9<0){print $3,$8,$8-$9+1,$1} else if($9>0){print $3,$4,$4+$9-1,$1}}' %s>%s
""" % (sam_file, sample_amplicon_bed)
cmd2 = """
%s coverage -b %s -a %s -f 0.96 -r >%s
""" % (bedtools, sample_amplicon_bed, amplicons_with_primers, sample_amplicon_depth)
参考资料
- Biostar http://www.biostars.org/p/16121/
- http://www.plob.org/2012/09/26/3748.html
- 官网 http://bedtools.readthedocs.org/en/latest/content/tools/genomecov.html
- Biostar http://www.biostars.org/p/5165/ (里面有很多中方法,非常不错的一个帖子)
