【5.3】blast简介及参数设置
blast几乎是无人不知的序列比对的工具,我说的是对学生物的同学,特别是搞bioinformatics。该工具一种是在线的( http://blast.ncbi.nlm.nih.gov/Blast.cgi ),一种是本地的(Local BLAST,本地版blast)
假设你有几条序列,想要看看这条序列是不是在某个物种中有类似的序列,或者你想在整个蛋白或者核酸数据库中搜索对应的同源序列,一般采用在线BLAST,使用ncbi的服务器,很快捷,界面也很美观。对于大量序列或者是自定义的数据库,一般采用本地BLAST,效率很更高。想想成千上万次手动提交序列,简直是不可能的。而且短时间内不停对服务器发起链接说不定会被ncbi ban掉(我就被ban了好几次,不过不是因为blast)。或者你想使用自己独特的数据库,不想使用什么NR,NT等等数据库的话,你就需要构建自己本地的数据库了一、Blast简介
- 相似的序列(similarity)可以预测同源(homology)
- 同源的序列来预测功能。这就是blast的根本目的。
2.局部相似性和整体相似性
序列比对的基本思想,是找出检测序列和目标序列的相似性。比对过程中需要在检测序列或目标序列中引入空位,以表示插入或删除(图3.1)。序列比对的最终实现,必须依赖于某个数学模型。不同的模型,可以从不同角度反映序列的特性,如结构、功能、进化关系等。很难断定,一个模型一定比另一个模型好,也不能说某个比对结果一定正确或一定错误,而只能说它们从某个角度反映了序列的生物学特性。此外,模型参数的不同,也可能导致比对结果的不同。
序列比对的数学模型大体可以分为两类,一类从全长序列出发,考虑序列的整体相似性,即整体比对;第二类考虑序列部分区域的相似性,即局部比对。局部相似性比对的生物学基础是蛋白质功能位点往往是由较短的序列片段组成的,这些部位的序列具有相当大的保守性,尽管在序列的其它部位可能有插入、删除或突变。此时,局部相似性比对往往比整体比对具有更高的灵敏度,其结果更具生物学意义。
区分这两类相似性和这两种不同的比对方法,对于正确选择比对方法是十分重要的。应该指出,在实际应用中,用整体比对方法企图找出只有局部相似性的两个序列之间的关系,显然是徒劳的;而用局部比对得到的结果也不能说明这两个序列的三维结构或折叠方式一定相同。BLAST和FastA等常用的数据库搜索程序均采用局部相似性比对的方法,具有较快的运行速度,而基于整体相似性比对的数据库搜索程序则需要超级计算机或专用计算机才能实现。
二、程序的使用
2.1 安装程序
在 ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ 下载ncbi-blast-2.2.28+-x64-linux.tar.gz
解压到用户的主目录(/sam)下(该目录位置你可以自己设定,跟后面统一即可),把解压后的文件夹重新命名为blast,则BLAST+的所有程序在目录/sam/blast/bin下。
添加环境变量
打开终端(Terminal),切换为root用户,执行
sudo vi /etc/profile
在最末尾添加
export PATH=”/sam/blast/bin:$PATH”
保存退出。
或直接找到/etc/profile这个文件,在最末尾添加
export PATH=”/sam/blast/bin:$PATH”
此处若成功,在终端则执行blastn -version会出现版本信息。
我的例子:
wget -c ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-2.7.1+-x64-linux.tar.gz
tar -xzvf ncbi-blast-2.7.1+-x64-linux.tar.gz
vim /etc/profile
#添加:
#Blast
export PATH=/home/sam/software/ncbi-blast-2.7.1+/bin:$PATH
source /etc/profile
因为是blast+ ,没有blastall的formatdb。同时blast后期已不再维护了呢。
在目录/sam/blast下新建一个文件夹,命名为db 在/sam下新建一个文件,命名为.ncbirc 在文件中添加内容
[BLAST]
BLASTDB=/sam/blast/db
(我后来找不到路径的时候我在/home/sam也同样建立了这样的一个文件,如果你不是在/home/sam下建立数据库,你得同时在这个地方建立./nvbirc的文件)
主要是blast识别~/.ncbirc文件,从该文件处得知数据库的位置,这个文件很重要!!!
2.2 下载FASTA格式的数据库
2.2.1自己的数据库
A. 如果是要做自己的数据库,要有自己的fasta文件,使用blast软件中所带的程序,将fasta文件 转变为blast的数据库文件:
B. /db下的数据库更小,下载更快;可以生成FASTA格式;在/sam/blast/bin/update_blastdb.pl可以用来升级数据库;不用formatdb数据库;各种ID可以进入这个数据库
2.2.2公共数据库
载地址 ftp://ftp.ncbi.nlm.nih.gov/blast/db/ 里面可以看到,有很多个分开的文件,但是其中FASTA也有很多个fasta格式的数据库,是一个完整的包,这个文件和上一节文件夹里面的数据有啥区别呢
Contents of the /blast/db/ directory
The pre-formatted BLAST databases are archived in this directory. The
name of these databases and their contents are listed below.
+----------------------+-----------------------------------------------+
|File Name | Content Description |
+----------------------+-----------------------------------------------+
/FASTA | subdirectory for FASTA formatted sequences
README | README for this subdirectory (this file)
env_nr.*tar.gz | Environmental protein sequences
env_nt.*tar.gz | Environmental nucleotide sequences
est.*tar.gz | volumes of the formatted est database| from the EST division of GenBank, EMBL, | and DDBJ
est_human.tar.gz | alias and mask files for human subset of the est
est_mouse.tar.gz | alias and mask files for mouse subset of the est
est_others.tar.gz | alias and mask files for non-human and non-mouse | subset of the est database | These alias and mask files need all volumes of | est to function properly.
gss.*tar.gz | volumes of the formatted gss database| from the GSS division of GenBank, EMBL, and | DDBJ
htgs.*tar.gz | volumes of htgs database with entries | from HTG division of GenBank, EMBL, and DDBJ
human_genomic.*tar.gz | human RefSeq (NC_######) chromosome records| with gap adjusted concatenated NT_ contigs
nr.*tar.gz | non-redundant protein sequence database with | entries from GenPept, Swissprot, PIR, PDF, PDB, | and NCBI RefSeq
nt.*tar.gz | nucleotide sequence database, with entries| from all traditional divisions of GenBank, | EMBL, and DDBJ excluding bulk divisions (gss, | sts, pat, est, and htg divisions. wgs entries | are also excluded. Not non-redundant.
other_genomic.*tar.gz | RefSeq chromosome records (NC_######) for | organisms other than human
pataa.*tar.gz | patent protein sequence database
patnt.*tar.gz | patent nucleotide sequence database| The above two databases are directly from | USPTO or from EU/Japan Patent Agencies via | EMBL/DDBJ pdbaa.*tar.gz | protein sequences from pdb protein structures, | its parent database is nr.
pdbnt.*tar.gz | nucleotide sequences from pdb nucleic acid | structures, its parent database it nt. They are | NOT the protein coding sequences for the | corresponding pdbaa entries.
refseq_genomic.*tar.gz | NCBI genomic reference sequences
refseq_protein.*tar.gz | NCBI protein reference sequences
refseq_rna.*tar.gz | NCBI Transcript reference sequences
sts.*tar.gz | Sequences from the STS division of GenBank, EMBL, | and DDBJ
swissprot.tar.gz | swiss-prot sequence databases (last major update), | its parent database is nr.
taxdb.tar.gz | Additional taxonomy information for the formatted | database (contains common and scientific names)
wgs.*tar.gz | volumes for whole genome shotgun sequence assemblies | for different organisms
+----------------------+-----------------------------------------------+
Contents of the /blast/db/FASTA directory
This directory contains FASTA formatted sequence files. The file names
and database contents are listed below. These files are now archived
in .gz format and must be processed through formatdb before they can be
used by the BLAST programs.
+-----------------------+-----------------------------------------------+
|File Name | Content Description |
+-----------------------+-----------------------------------------------+
alu.a.gz | translation of alu.n repeats
alu.n.gz | alu repeat elements
drosoph.aa.gz | CDS translations from drosophila.nt
drosoph.nt.gz | genomic sequences for drosophila
env_nr.gz* | Environmental protein sequences
env_nt.gz* | Environmental nucleotide sequences
est_human.gz* | human subset of the est database (see Note 1)
est_mouse.gz* | mouse subset of the est database
est_others.gz* | non-human and non-mouse subset of the est
database
gss.gz* | sequences from the GSS division of GenBank, | EMBL, and DDBJ
htg.gz* | htgs database with high throughput genomic | entries from the htg division of GenBank, | EMBL, and DDBJ
human_genomic.gz* | human RefSeq (NC_######) chromosome records | with gap adjusted concatenated NT_ contigs
igSeqNt.gz | human and mouse immunoglobulin variable region nucleotide | sequences
igSeqProt.gz | human and mouse immunoglobulin variable region protein | sequences
mito.aa.gz | CDS translations of complete mitochondrial | genomes
mito.nt.gz | complete mitochondrial genomes
month.aa.gz | newly released/updated protein sequences
(See Note 2)
month.est_human.gz | newly released/updated human est sequences
month.est_mouse.gz | newly released/updated mouse est sequences
month.est_others.gz | newly released/updated est other than | human/mouse
month.gss.gz | newly released/updated gss sequences
month.htgs.gz | newly released/updated htgs sequences
month.nt.gz | newly released/updated sequences for the nt
database
nr.gz* | non-redundant protein sequence database with| entries from GenPept, Swissprot, PIR, PDF, | PDB, and RefSeq
nt.gz* | nucleotide sequence database, with entries | from all traditional divisions of GenBank, | EMBL, and DDBJ excluding bulk divisions| (gss, sts, pat, est, htg divisions) and wgs | entries. Not non-redundant.
other_genomic.gz* | RefSeq chromosome records (NC_######) for | organisms other than human
pataa.gz* | patent protein sequence database
patnt.gz* | patent nucleotide sequence database | The above two dbs are directly from USPTO | of from EU/Japan Patent Agency via EMBL/DDBJ
pdbaa.gz* | protein sequences from pdb protein structures
pdbnt.gz* | nucleotide sequences from pdb nucleic acid | structures. They are NOT the protein coding | sequences for the corresponding pdbaa entries.
sts.gz* | database for sequence tag site entries
swissprot.gz* | swiss-prot database (last major release)
vector.gz | vector sequence database (See Note 3)
wgs.gz* | whole genome shotgun genome assemblies
yeast.aa.gz | protein translations from yeast genome
yeast.nt.gz | yeast genomes.
+-----------------------+-----------------------------------------------+
FASTA格式的数据库,需要写入,便于搜索
cd /sam/balst/db
formatdb -i input_db -p F -o T for nucleotide
formatdb -i input_db -p T -o T for protein
-i 输入需要格式化的源数据库名称 Optional
-p 文件类型,是核苷酸序列数据库,还是蛋白质序列数据库
T – protein F - nucleotide [T/F] Optional default = T
-a 输入数据库的格式是ASN.1(否 则是FASTA)
T - True, F - False. [T/F] Optional default = F
-o 解析选项
T - True: 解析序列标识并且建立目录 F - False: 与上相反
[T/F] Optional default = F
注:1 因为我选择的是/db下有许多个文件夹组成的某一个数据库,所以就没有写入这个过程。
2.2.2 在/db下下载的很多个数据库,比如nr00,nr01,nr02……解压缩后要放到一个文件夹里面,用数据库的时候,
blast -db /sam/blast/db/nr/nr
我在这个问题上费了很久,刚开始是选择什么样的数据库,后来是下载的nr分开的数据库如何整合到一起。其实运行的是那个唯一不带编号的Nr文件。
2.2.3 以下的命令是网上找到的,针对FASTA格式的,但是我没用过,把它放在这上面,供大家参考吧
建立BLAST+可用的数据库:
打开终端(Terminal),切换到/sam/blast/db目录下,执行:
makeblastdb –in nr -parse_seqids -hash_index -dbtype prot
或者是对某个fasta文件进行处理:其中的makeblastdb在bin文件夹里面。
makeblastdb -in db.fasta -dbtype prot -parse_seqids -out dbname
参数说明:
-in:待格式化的序列文件
-dbtype:数据库类型,prot或nucl
-out:数据库名
-parse_seqids 根据序列ID来分裂
具体参数的设置可以参见makeblastdb -help
3.选择程序
3.1 常规程序
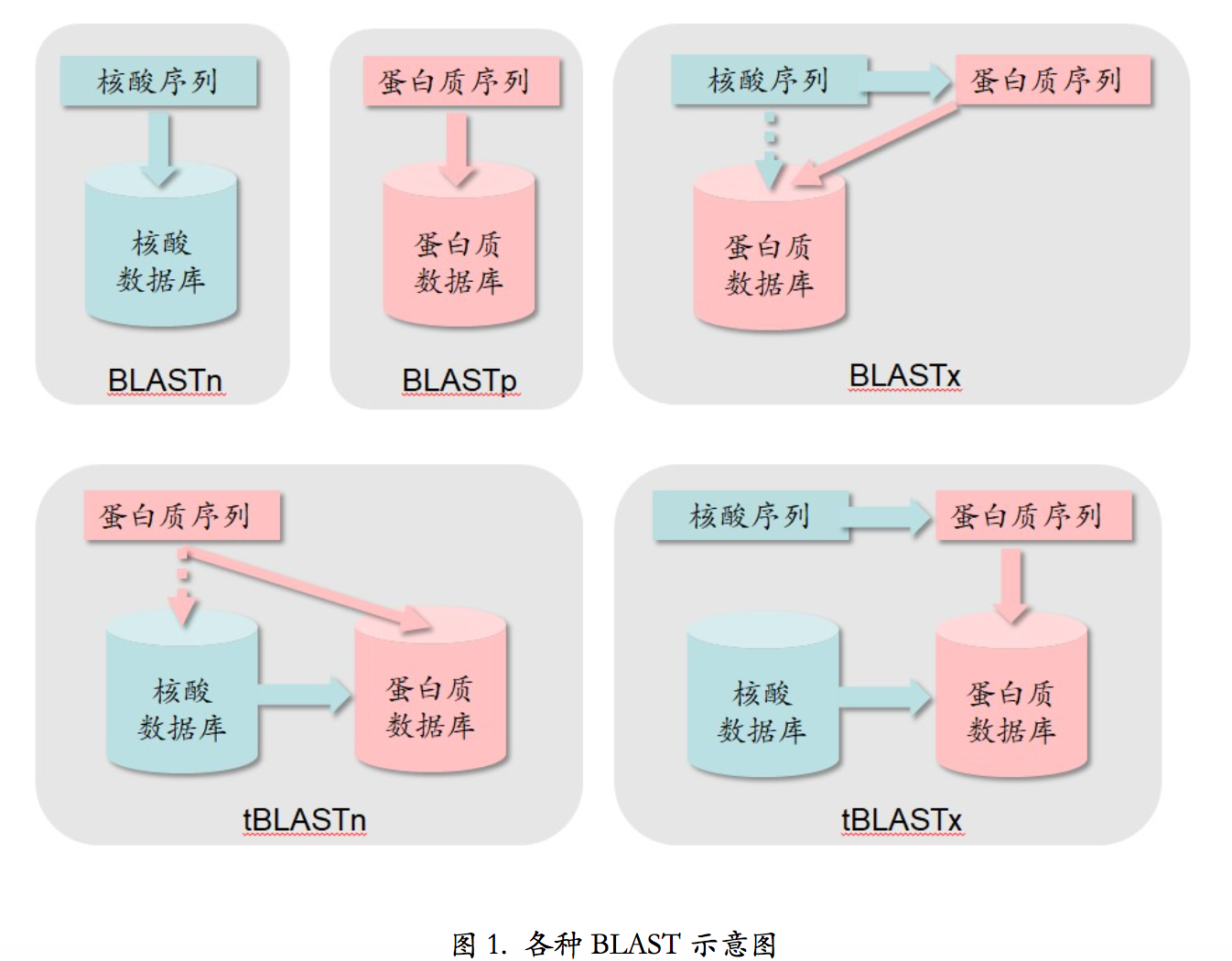
- blastp:用蛋白质序列搜索蛋白质序列库
- blastn:用核酸序列搜索核酸库
- blastx:核酸序列对蛋白质库的比对,核酸序列在比对之前自动按照六个读码框翻译成蛋白质序列。BLASTx 是将核酸序列按 6 条链翻译成蛋白质序列后搜索蛋白质序列数据库。为什么是 按 6 条链翻译?在无法得知翻译起始位点在情况下,翻译可能是从第一个碱基开始,三个三 个的往后翻译,也可能是从第 2 个碱基开始,也可能从第 3 个碱基开始。另外还有可能是从 这条链的互补链上开始,这样又有三个可能的开始位置,加起来一共会产生 6 条可能被翻译 出来的蛋白质序列。这 6 条中有些是真实存在的,有些是不存在,但是谁真谁假我们无从知 晓,所以 6 条序列都要到数据库中去搜索一下试试。接下来的问题是,既然是核酸序列,为 什么不做 BLASTn 直接到核酸数据库里去搜索,而是要到蛋白质数据库里搜索呢?我们说这 样做是有意义的,比如,从核酸序列数据库里找不到跟你手里这条核酸序列相似的序列,或 找到了相似的序列但这些找到的序列无法提供有意义的注释信息。这时,就可以去蛋白质数 据库试试,看看这条核酸序列的翻译产物能不能从蛋白质数据库里找到相似的序列以及有意 义的注释信息。或者说,你不是想找跟你这条核酸序列相似的核酸序列,而是想找跟你这条 核酸序列编码蛋白质相似的蛋白质序列,这时就要做 BLASTx。
- tblastn:蛋白质序列对核酸库的比对,核酸库中的序列按照六个读码框翻译后与蛋白质序列进行比对搜索。反之,当你不是想找跟你手上这条蛋白质序列相似的蛋白质序列,而是想找跟编码这条 蛋白质序列的核酸序列相似的核酸序列的时候,就要做 tBLASTn。tBLASTn 是用蛋白质序列 搜核酸序列数据库,核酸数据库中的核酸序列要按 6 条链翻译成蛋白质序列后再被搜索。你 可能要问了,核酸数据库里不是已经注释了某条核酸序列能够翻译成什么蛋白质序列吗?为 什么还要把这些序列可能翻译出来的 6 条蛋白质序列都翻译出来搜索呢?我们说,你看到的 是已经注释的,还有没注释的呢!就算是已经注释的,你看到的也只是已经研究出来的成果, 还有没研究出来的呢!别忘了,基因可以重叠,注释上说某段 DNA 序列可以编码某个蛋白,但是可能某个未被发现的基因也用到了这段 DNA 序列。而你要搜索的这个蛋白质序列可能 刚好就是这个未被发现的基因的翻译产物。这样就必须把核酸序列所有可能的翻译产物都翻 译出来,才能搜索得到。
- tblastx:核酸序列对核酸库在蛋白质质级别的比对,两者都在搜索之前翻译成为蛋白质质进行比对。上述研究方法运用到极限就是 tBLASTx。它是将核酸序列按 6 条链翻译成蛋白质序列后 搜索核酸序列数据库,核酸数据库中的所有核酸序列也要按 6 条链翻译成的蛋白质序列后再 被搜索。这样用 BLASTn 搜不着的,用 tBLASTx 就能搜着了。
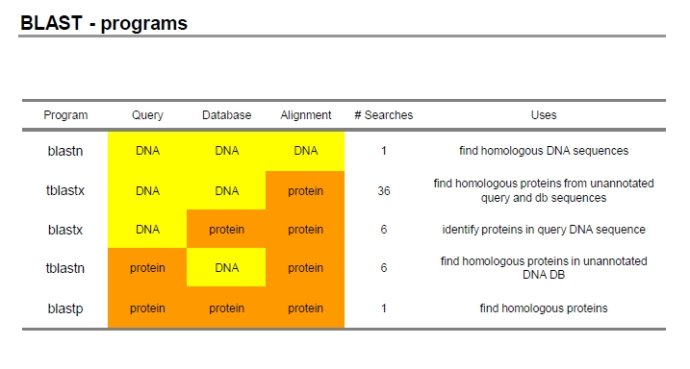
(为什么会是6呢,因为DNA在翻译为蛋白质的时候移位,一个aa由三个碱基构成,所以可以移动3个,同时又是因为正反链,所以会有6种情况。)
氨基酸替代矩阵
PAM=Point Accepted Mutations 相近的序列比对得出的结论
BLOSUM = Blocks Amino Acid Substitution Matrices,来自BLOCKS database ([http://blocks.fhcrc.org/](http://blocks.fhcrc.org/))
The numbers mean different things depending on the particular matrix. For the
PAM matrix, the high number matrices are better for more divergent alignments, while the
opposite is true for the BLOSUM matrices.
假如我们找距离比较远的序列,我们可以用PAM更大数目的举证或者是BLOSUM更小的矩阵
3.2 Position-Specific Iterated (PSI)-BLAST:
3.3 PHI-BLAST
PHI-BLAST 和 PSI-BLAST 不同,PSI-BLAST 是撒大网搜索,而 PHI-BLAST 则是精准搜 索。PHI 是 Pattern-Hit Initiated 首字母缩写,中文是模式识别。PHI-BLAST 能找到与输入序 列相似的并符合某种特征模式的蛋白质序列。模式 Pattern 是对特征的描述。通俗的说,比 如某个会议的会场服务人员都符合这样一个特定的模式:短发戴眼镜穿白色 T 恤的女生。 换到生物领域,比如发生 N 糖基化位点的序列都符合这样一个特定的模式:发生糖基化的 天冬酰胺,后面一定紧跟一个脯氨酸以外的氨基酸,再紧跟丝氨酸或者苏氨酸,再紧跟一个 脯氨酸以外的氨基酸。
特定模式可通过正则表达式来表述。所谓正则表达式就是这句话的一个简约的规范性字 符书写法。发生 N 糖基化位点的序列符合的特定模式翻译成正则表达式为 N{P}[ST]{P}。 其中,N 是天冬酰胺,P 是脯氨酸,S 是丝氨酸,T 是苏氨酸。{}代表除大括号里的氨基酸 以外的任意氨基酸,[]代表中括号中的任意一个氨基酸。得知这些符号的含义之后,这个 正则表达式就很容易读懂了。PHI-BLAST 可以根据给入的正则表达式对搜索到的相似序列 进行模式匹配,符合正则表达式的才会被作为结果输出。
我们再来熟悉一下正则表达式,{}代表除什么以外,[]代表其中之一,x 代表任意字 母,(3,7)代表 3 到 7 个某字符。那么正则表达式{L}GEx[GAS][LIVM]x(3,7)的意思是, 除 L 以外的任意一个字母,紧接 G,再紧接 E,再接一个任意字符,之后是 GAS 中的任意 一个,再接 LIVM 中的任意一个,最后再接 3 到 7 个任意字符。据此,VGEAAMPRI 这条序 列是符合这条正则表达式的,而 VGEAAYPRI 这一条则不符合。这种序列特征模式可能代表 某个翻译后修饰的发生位点,也可以代表一个酶的活性位点,或者一个蛋白质家族的结构域、 功能域等有重要意义的地方。
在 NCBI BLAST 工具的输入页面,当算法选择了 PHI-BLAST 之后,会自动出现模式输 入框(图 1)。输入正则表达式 S[IVFL]TPS(2)(含义为:一个 S 后面紧接 IVFL 中的任意 一个字母,再接 T,再接 P,再接两个 S)。这次搜索找到的相似序列中,只有符合该模式的 才会被作为结果返回。

3.4 其他的blast
除了前面讲的这三种 BLAST,NCBI 还开发了一个 SMART-BLAST(聪明的 BLAST)。 SMART-BLAST 可谓是标准 BLASTp 的简约强化版。操作极其简单,简单到只需要输入序列。
SMART-BLAST 虽然操作简单,但返回的结果却并不简单(图 2)。它包括数据库中与输 入序列最相似的三条序列,以及研究的最透彻的物种中可以展现一定进化关系的两条相似序 列。图 2 中黄色的是你输入的序列,绿色的是研究的最好的模式物种中与你输入序列相似的 序列。旁边还直接给出了这三条序列的系统发生树。总之,SMART-BLAST 可以帮你从一大 堆结果中挑选出你最想要的。如果你很懒,或者你很茫然,可以试试这个聪明的 BLAST。
你在实际操作中也许会发现,NCBI 的网站好慢啊!没关系,我们前面说过,各大知名 数据库网站都提供 BLAST 搜索工具,而且可以跨平台搜索(表 1)。因此,可以利用时差, 挑美国人睡觉的时候上 NCBI BLAST,挑欧洲人睡觉的时候去 Expasy 或者 Uniprot BLAST, 亚洲人睡觉的时候去 DDBJ BLAST。
| 位置 | 服务器 | 网址链接 |
|---|---|---|
| USA | NCBI | http://www.ncbi.nlm.nih.gov/BLAST |
| Europe | ExPASy | http://web.expasy.org/blast |
| Europe | Uniprot | http://www.uniprot.org/blast/ |
| Japan | DDBJ | http://blast.ddbj.nig.ac.jp |
此外,除了 BLAST,还有其他的数据库相似性搜索工具,比如 WU-BLAST。WU 代表 Washington University,它比 BLAST 更灵敏,在插入空位的算法上更灵活。再有 SSEARCH, 它有点儿慢,但是比 BLAST 更准确。FASTA 也是一个搜索工具,它也是有点儿慢,但是对 于 DNA 序列的比较比 BLAST 更准确,尤其适合短的序列。最早被 FASTA 使用的序列格式 就叫 FASTA 格式,并沿用至今。这就是大于号开头的 FASTA 格式的由来。还有一种叫 BLAT, 用于小的序列在大基因组中的搜索。
4.使用程序:
/sam/blast/bin/blastp -query assembly.orfs.hmm.faa -db /sam/blast/db/refseq_protein/refseq_protein -evalue 1e-5 -num_threads 10 -max_target_seqs 5 -outfmt 5 -out assembly.orfs.hmm.blast.xml
-query 输入序列
-db 选择的数据库
-evalue 这个就不说了
-num_threads 服务器使用的cpu个数
-max_target_seqs设置最多的目标序列匹配数
-out输出的文件名
-remote 如果加上这个参数,就是使用远程比对,再有互联网的情况下,连接到NCBI的服务器来进行远程比对,然后返回到本地,从而不消耗本地的资源,设置了该参数,则-num_threads无效
-perc_identity 相似度,默认是大于0就显示,如果要显示大于95%,这后面接95
-outfmt
5代表xml格式,这个格式的信息最全,6代表table格式,该格式的结果以table的形式给出,清晰易懂,7代表有注释行的table格式,比6多了一些#开头的注释行。
<strong> 注意文件或程序的路径,如果没有cd到相应的位置的话,就需要在命令中注明路径</strong>
如果你对比对后的结果有自己的要求的话,建议xml用5这个格式的输出,然后用脚本来提取,我之前用python写过一个这样的脚本:blast结果的提取
“7 qacc sacc evalue length pident” :这个是新BLAST+中最拉风的功能了,直接控制输出格式,不用再用parser啦, 7表示带注释行的tab格式的输出,可以自定义要输出哪些内容,用空格分格跟在7的后面,并把所有的输出控制用双引号括起来,其中qacc查询序列的acc,sacc表示目标序列的acc,evalue即是e值,length即是匹配的长度,pident即是序列相同的百分比,其他可用的特征(红色字体)如下:
Formatting options
# 通过 -outfmt 参数来定义各种形式的输出
alignment view options:
0 = pairwise
1 = query-anchored showing identities,
2 = query-anchored no identities,
3 = flat query-anchored, show identities,
4 = flat query-anchored, no identities,
5 = XML Blast output,
6 = tabular,
7 = tabular with comment lines,
8 = Text ASN.1,
9 = Binary ASN.1
10 = Comma-separated values
Options 6, 7, and 10 can be additionally configured to produce
a custom format specified by space delimited format specifiers.
The supported format specifiers are:
qseqid means Query Seq-id
qgi means Query GI
qacc means Query accesion
sseqid means Subject Seq-id
sallseqid means All subject Seq-id(s), separated by a ';'
sgi means Subject GI
sallgi means All subject GIs
sacc means Subject accession
sallacc means All subject accessions
qstart means Start of alignment in query
qend means End of alignment in query
sstart means Start of alignment in subject
send means End of alignment in subject
qseq means Aligned part of query sequence
sseq means Aligned part of subject sequence
evalue means Expect value
bitscore means Bit score
score means Raw score
length means Alignment length
pident means Percentage of identical matches
nident means Number of identical matches
mismatch means Number of mismatches
positive means Number of positive-scoring matches
gapopen means Number of gap openings
gaps means Total number of gaps
ppos means Percentage of positive-scoring matches
frames means Query and subject frames separated by a '/'
qframe means Query frame
sframe means Subject frame
When not provided, the default value is:
'qseqid sseqid pident length mismatch gapopen qstart qend sstart send
evalue bitscore', which is equivalent to the keyword 'std'
Default = `0'
例子:
cmd = """blastp -query %s -db %s -evalue 1000 -num_threads 40 -max_target_seqs 4000 -outfmt "6 qaccver saccver pident length mismatch gapopen qstart qend sstart send evalue bitscore qlen slen length pident nident" -out %s""" % (input_fa,input_db,output_file)
调用blastn合作加-help参数可以打印出下面详细的帮助信息 blastn -help。
-outfmt 0
这个格式的输出跟 NCBI官网上看到的一样,包含序列比对情况概况和序列比对位置分析
因为 -max_target_seqs 不适用于输出格式为0,1,2,3。但我们可以通过-num_descriptions 指定显示出来序列比对概况的序列数,通过 -num_alignments指定序列比对位置详情的比对结果个数
-outfmt 1
跟0的区别在于,没有了score、gap等的概况,query和hit的序列名字都用真是的名字
-outfmt 2
2跟1的区别在于,不相似的残基没有用空格表示了
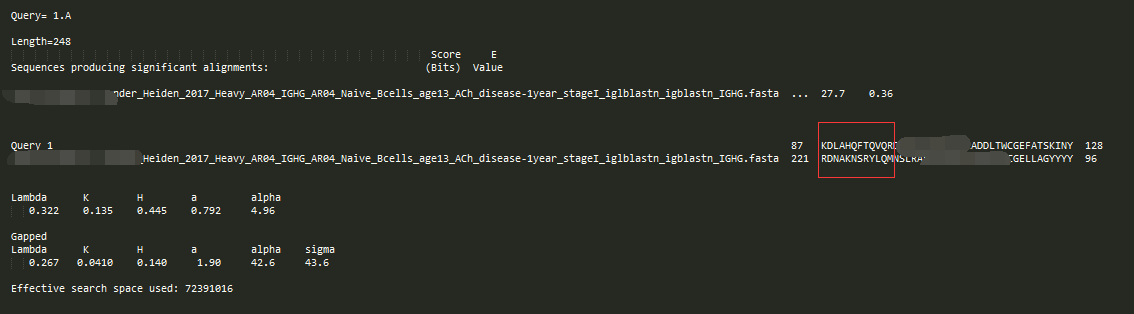
-outfmt 3
2和1的区别在于,对query插入的处理显示,
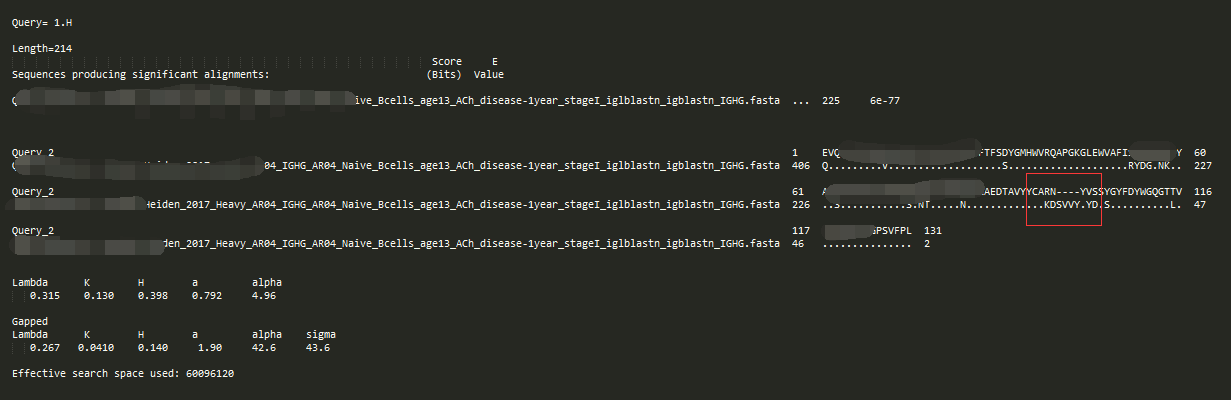
-outfmt 4
4跟3的区别在于,不相似的残基没有用空格表示了
总结:这么看来,我更喜欢0作为变现形式。
4 手动注释–tblastn
我们从宏基因组中聚类出我们感兴趣的微生物物种的基因组后,想要预测其潜在的新的代谢途径的时候,一般会有两种策略:
- 对该基因组进行注释,获得所有的注释结果,然后对注释结果的解读;
- 通过看文献,搜集一些感兴趣的相关代谢途径的关键基因,把这些关键基因的蛋白序列给下下来,然后在你的基因组里面找看有没有相关的基因。第一种策略的注释之前的博文零星的有一些介绍,下面主要讲一下第二种策略所涉及到的一个方法—-tblastn.
案例:
$ cd /sam/uncltured/contig7/c3000/syn/genome2/tblastn
$ makeblastdb -in genome2.fasta -dbtype nucl -title syn1 -parse_seqids -out syn1 -logfile syn1.log #这一步是将的基因组作为数据库
$ /sam/blast/bin/tblastn -query /sam/syn/alkylsuccinateSynthase_α-subunit_assA.fasta -db syn1 -evalue 1e-5 -num_threads 60 -outfmt 5 -out assA.xml #比对,得到的是一个xml文件
$ /sam/blast/bin/tblastn -query /sam/syn/alkylsuccinateSynthase_α-subunit_assA.fasta -db syn1 -evalue 1e-5 -num_threads 60 -outfmt 7 -out assA #比对,得到的是一个table文件
其实-outfmt 5得到的结果就是一个比较详尽的结果,-outfmt 7得到的是一个table文件。
# TBLASTN 2.2.28+
# Query: gi|299800799|gb|ADJ51097.1| alkylsuccinate synthase [Desulfoglaeba alkanexedens ALDC]
# Database: syn1
# Fields: query id, subject id, % identity, alignment length, mismatches, gap opens, q. start, q. end, s. start, s. end, evalue, bit score
# 9 hits found
gi|299800799|gb|ADJ51097.1| 109 69.50 341 104 0 365 705 4 1026 5e-169 494
gi|299800799|gb|ADJ51097.1| 226 68.48 257 81 0 449 705 773 3 4e-118 395
gi|299800799|gb|ADJ51097.1| 226 73.17 82 22 0 290 371 1007 762 2e-32 132
gi|299800799|gb|ADJ51097.1| 839 31.61 794 504 19 77 858 13150 10850 7e-91 316
gi|299800799|gb|ADJ51097.1| 292 87.23 47 6 0 812 858 1 141 2e-20 90.1
gi|299800799|gb|ADJ51097.1| 900 87.23 47 6 0 812 858 111138 110998 2e-19 90.5
gi|299800799|gb|ADJ51097.1| 201 48.44 64 33 0 36 99 192 1 9e-13 68.9
gi|299800799|gb|ADJ51097.1| 322 46.88 64 34 0 36 99 192 1 2e-12 67.4
gi|299800799|gb|ADJ51097.1| 1072 60.00 35 14 0 824 858 7642 7746 2e-06 48.1
# BLAST processed 1 queries
#当然输入序列也可以使好多个蛋白序列合并的fasta文件,例如,我对alkylsuccinateSynthase感兴趣,我可以到ncbi中把相关的基因的蛋白序列都给下下来,放到一个文件里面。
四、讨论
4.1 短序列的比对
blastn -task blastn-short
4.2 其他
1.我在下载蛋白序列的时候,发现有的序列长度为800个aa,有的仅为200个aa的parital,为什么partial也能放到数据库中呢?
2.比对的结果,如何设置阈值,判断结果中跟输入序列有很高的同源性呢?
3.fastacmd 提取序列
target_seq = '%s -d %s -p F -s "lcl|%s" -L%s,%s' % (
fastacmd, genome, chrom, start, end)
4.设置比对序列覆盖的程度
-qcov_hsp_perc <real, 0..100=""> Percent query coverage per hsp
五、报错
5.1 报错1
makeblastdb的时候
T0 "/home/coremake/release_build/build/PrepareRelease_Linux64-Centos_JSID_01_350334_130.14.22.10_9008__PrepareRelease_Linux64-Centos_1481139955/c++/compilers/unix/../../src/objects/seq/../seqloc/Seq_id.cpp", line 1911: Error: ncbi::objects::CSeq_id::x_Init() - Unsupported ID type 1196094
Dose any
这个跟序列的名字有关。
解决办法:
makeblastdb -in db.fasta -dbtype prot -parse_seqids -out dbname
改为(不在根据序列ID来编号)
makeblastdb -in db.fasta -dbtype prot -out dbname
5.2 报错2:BLAST Database error: Error: Not a valid version 4 database.
(metawrap-env) [sam@g01 21]$ blastn -task megablast -num_threads 40 -db /data/database/metawrap/NCBI_nt/nt -outfmt '6 qseqid qstart qend qlen sseqid staxids sstart send bitscore evalue nident length' -query test.fasta
BLAST Database error: Error: Not a valid version 4 database.
解决办法:
blast的版本和数据库的版本不一致
wget -c ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-2.10.1+-x64-linux.tar.gz
tar xzvfm ncbi-blast-2.10.1+-x64-linux.tar.gz
/data/software/blast/ncbi-blast-2.10.1+/bin
export PATH=/data/software/blast/ncbi-blast-2.10.1+/bin:$PATH
参考资料
