【7.2】使用 MinHash 快速估计基因组和宏基因组距离--Mash
Mash 扩展了 MinHash 降维技术,包括成对变异距离和P值显着性检验,从而实现对海量序列集合的高效聚类和搜索。Mash 将大序列和序列集缩减为小的、有代表性的草图,从中可以快速估计全局突变距离。
代码: https://github.com/marbl/mash
Mash 为序列比较提供了两个基本函数:sketch和dist。
- sketch的函数转换序列或序列集合到一个最小哈希草图(MinHash sketch)(图 1)。
- DIST函数比较两个草图和返回的Jaccard指数的估计值(共用的k聚体的即部分),一个P值和Mash距离,其估计序列突变的速率下一个简单的进化模型[ 22 ](见“方法”)。
由于 Mash 仅依赖于比较长度k子串或 k-mers,输入可以是全基因组、宏基因组、核苷酸序列、氨基酸序列或原始测序读数。每个输入都被简单地视为取自某些已知字母表的 k-mer 的集合,从而允许许多应用。
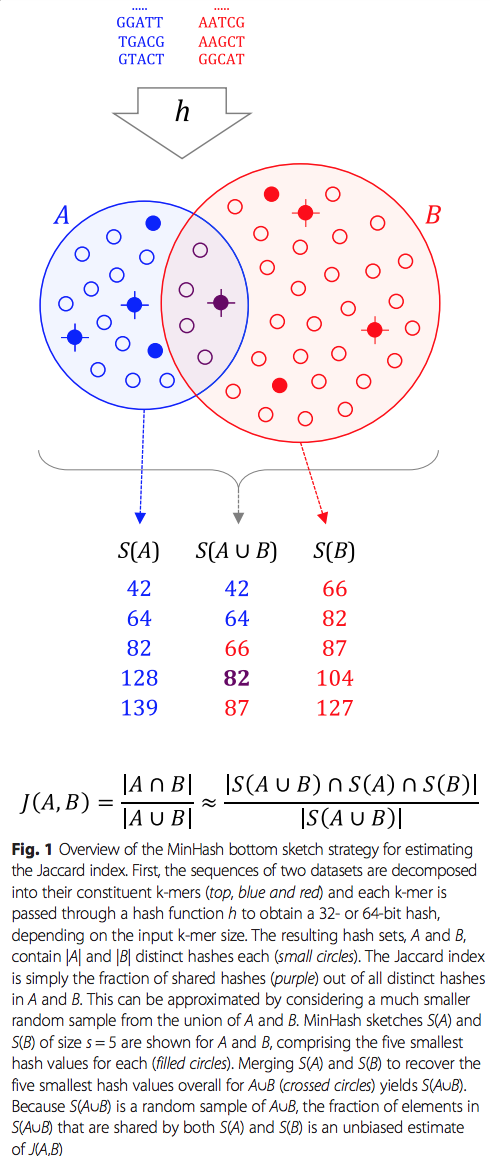
细心的读者可能注意到了,以上的计算中做了一个随机抽样的近似,这也是Mash可以大大节省资源消耗的一个关键,不难推论,抽样的群体越大,得到的估计值越接近真实值,遗憾的是,这样耗时也会相应增加,文章的实验结论也印证了这一点。
参考资料
- Mash: fast genome and metagenome distance estimation using MinHash。 https://link.springer.com/article/10.1186/s13059-016-0997-x
药企,独角兽,苏州。团队长期招人,感兴趣的都可以发邮件聊聊:tiehan@sina.cn
![]() 个人公众号,比较懒,很少更新,可以在上面提问题,如果回复不及时,可发邮件给我: tiehan@sina.cn
个人公众号,比较懒,很少更新,可以在上面提问题,如果回复不及时,可发邮件给我: tiehan@sina.cn
