【3.5.1】系统进化树
一、进化的故事
1.1 拉马克与用进废退
故事要从一位俄罗斯的科学家杜布赞斯基说起。1973 年杜布赞斯基说了一句非常著名 的话:Nothing in Biology Makes Sense Except in the Light of Evolution(如果生物学没有了进化, 那么一切都将黯然无光)。你可能觉得这话说得有点儿夸张,但是结合他说此话的目的,就 可以理解了。因为对于杜布赞斯基所处的宗教社会而言,造物主理论和智慧设计论始终是社 会的主流理论。他在《美国生物教师》这样一本刊物上说出这样一句话的用意可想而知。这 里我们不讨论宗教,我们只说科学。如果你不认可也没有关系,毕竟从古至今,对这个领域 的争论从来没有停止过。但是无论何时、无论何地我们都可以看到进化或以正面或以反面存 在着。特别是对于研究生命科学的我们,进化尤为重要。
说到进化论就不得不提一个人和他的理论,那就是大家都熟悉的达尔文和他的进化论。 但是在达尔文之前,就已经有一个人开始研究生物进化了。他才是进化研究的鼻祖,没有他 就没有达尔文。他就是拉马克!
拉马提出进废退理论。他说生物经常使用的器官会逐渐发达,不经常使用的器官会逐渐 退化。而且这种后天获得的性状是可以遗传的,因此生物可以把后天锻炼的成果遗传给下一 代。拉马克举了长颈鹿的例子。他说,长颈鹿原本脖子没那么长,但是为了吃到高处树叶不 断的伸长脖子,久而久之,他的脖子越伸越长,并且把长脖子这个后天获得的性状,遗传给 了下一代。再有鼹鼠一年到头的在地下打洞。因为地下漆黑一片,打洞只要鼻子闻,根本用 不着眼睛看,久而久之,都成了瞎子。再有鸟吃饭都不嚼直接咽,所以鸟嘴里都没牙,而哺 乳动物比如猫要撕咬食物,所以尖牙利齿。
拉马克主义提出后,生物界支持声和反对声此起彼伏。先来听听反对他的声音。德国科 学家魏斯曼做了一个实验。他把老鼠的尾巴都切断,之后再让没有尾巴的老鼠互相交配,生 出的下一代老鼠依然是有尾巴的。然后再把子代老鼠的尾巴切断之后交配,生出的下一代老 鼠依然是有尾巴的。实验一直重复至第 21 代,但是老鼠的尾巴依然长长的,一点儿都没短。 于是魏斯曼说拉马克是错的!
再来听听支持的声音。水生的雄蟾蜍都有一个黑色的趾垫,而陆生的没有。奥地利科学 家卡姆梅勒,强迫陆生的产婆蟾在水中生活。繁殖了几代之后就绝种了。但是在绝种之前, 产婆蟾的雄蟾蜍据称是长出了黑色的指垫,而且水中生活的这几代,趾垫越来越明显。虽然 终究因为不适应水生生活灭绝了,但是在趾垫这个问题上还是朝着拉马克主义发展的。为了 获得更多的科研经费,卡姆梅勒开始周游列国宣讲他的实验。后来遭到了英国科学家的质疑。 1926 年,美国自然历史博物馆和维也纳大学检查了他的产婆蟾标本,发现所谓长出的黑色 趾垫是用墨水涂上去的,属于学术造假。在被揭发造假行为一个月后,卡姆梅勒就开枪自杀 了,留下一封遗书,声称他是无辜的,是另外有人在他不知道的情况下造假。真相到底如何, 至今也无从知晓。
1.2 达尔文与自然选择
在大学里,达尔文对自然注意十分着迷。但是要以此作为事业的话,他需要习惯海上的颠簸。1835 年他抵达了加拉帕戈斯群岛,在那里,自然的独特本质撼动了达尔文的世界。一切始于加拉帕戈斯地雀,在这一列岛中, 每个岛上的地雀都长着独特的喙部。独特的喙部似乎都是为了适应不同的食物而生,不管是 种子、仙人掌、还是幼虫。达尔文认为这绝不可能是巧合,于是开始调查研究生物的适应性。 这项研究话费了 20 年才得以完成。然而他得出的结论却与当时盛行的神创论中关于生物起 源的一切观点相悖。达尔文认为,所有的生物物种都存在趋利的适应性变化。并且这些适应 性变化会通过一种他称之为“自然选择”的过程遗传下去。而大自然是数百万年间推动演变 进化的唯一力量。这个理念让达尔文成为了现代生物学之父。对于一个渴望成为牧师的人来 说,这个称谓也不赖。进化无时无刻不在发生,这一理念产生后,人们就想找到一种机制。 而这就是达尔文的贡献所在,也是他为何会花费如此长的时间进行思考、收集证据以及整理 入书。达尔文认为“物竞天择”意味着,存在一种最初的生物,之后通过某种方式得到了改 良。如果环境对你施加压力,压力有可能是捕食者的威胁或类似的情况,那些通过某种手段 生存下来,并且繁衍后代的生物,他们的后代也能生存下来,并且继续繁衍生息。因此,如 今我们所见到的动植物都拥有很强的适应性。他们所表现出来的适应性使得他们成功存活, 并继续繁衍下一代。[摘自 CCTV 记录片-你最想知道的科学]
所以现在你知道,如果让达尔文的长颈鹿和拉马克的长颈鹿 PK,谁能获胜?没错,截 至目前,我们更相信达尔文所说的。长颈鹿的脖子不是越伸越长,而是短脖子的长颈鹿都饿 死了。所以在这场 PK 中,后起之秀达尔文,完胜!
二、进化的基本概念
2.1 如何研究进化
拉马克和达尔文告诉我们,进化很重要!那么,究竟怎样才能研究进化呢?传统上有两 种研究进化的方法。一种是看死的,一种是看活的。看死的,也就是看化石,这是进化最直 接,最确凿的证据。假如我们可以获得某个物种自诞生之日起每隔一百年的所有完整化石, 那么我们就可以看出这个物种是怎么一点一点演变来的。但是非常可惜的是,生物化石很零 散,不完整。人类得到生物化石是个极其偶然的事件。
再来看第二种研究方法。第二种研究方法是利用比较形态学、比较解剖学和生理学等手 段,确定大致的进化框架。这种方法比第一种方法更容易实现。但是这种方法仅局限于大致 的框架,很多细节是存在争议的。只能是仁者见仁智者见智,因为用肉眼观察存在误差。还 有一个问题就是伦理学问题。拨开一只兔子要比拨开一个桔子难得多,而为了研究进化去拨 开很多很多人,听上去是个恐怖片儿!
两种传统方法都有局限性,所以今天我们研究的是分子进化。也就是利用软件,从分子 水平上构建物种的进化树。这里说的分子水平是指 DNA、RNA、以及蛋白质序列。通过前 几章的学习,我们知道随着测序技术的发张这种分子水平上的信息量大而丰富,获取十分便 捷。当然有优点自然就有缺点。缺点就是准确度依赖软件的优劣及参数的设置。随着相关软 件的不断升级,这个缺点也再逐渐被克服。
现在我们系统的了解一下分子进化。分子进化理论是 1962 年美国科学家 Linus Pauling 提出的。这种理论与传统研究方法的最大区别是,它研究的是 DNA、RNA 以及蛋白质序列 这些分子水平上的信息,而不是物种的外在特征。并且基于某一个特定的分子在不同物种中 的序列差异来构建系统发生树。此外,分子进化有两个基本的假设条件,只有接受这两个假 设,分子进化理论才能得以实施。第一、DNA、RNA 或蛋白质序列包含了物种的所有进化 史信息。第二是分子钟理论。这个理论说的是,一个特定基因或蛋白质的进化变异速度在不 同物种中是基本恒定的。所谓变异速度是指一定时间内不同碱基或氨基酸突变的个数。这个 进化变异速度被认为是恒定的,跟物种没有关系。所以,拿蛋白质来说,两个蛋白质在序列 上越相似,他们距离共同祖先就越近。分子钟理论是进化研究领域被普遍认可的理论,但是 至今也没有直接的证据证实。
2.2 不同的同源
同源(Homologs),相同来源。没错,但是它的确切定义是,来源于共同祖先的相似序 列为同源序列。也就是说,相似序列有两种,一种是来源于共同祖先的,那么他们可以叫同 源,另一种不是来源于共同祖先的,那么他们尽管相似也不能叫同源。第二种情况出现的概 率虽然低,但还是存在的,所以相似序列并不一定是同源序列。同源又分为三种,直系同源, 旁系同源和异同源。
直系同源(Orthologs)是指,来自于不同物种的由垂直家系,也就是物种形成,进化而 来的基因,并且典型的保留与原始基因相同的功能。也就是说,随着进化分支,一个基因进 入了不同的物种,并保留了原有功能。这时,不同物种中的这个基因就属于直系同源。
旁系同源(Paralogs)是指在同一物种中的来源于基因复制的基因,可能会进化出新的 但与原功能相关的功能来。基因复制产生了两个重复的基因,多出来的这个有几种命运,一 个是又丢了。复制出来发现没有用,又删了。另一种命运是演化出了新的功能。如果这个新 功能是往好的方向发展,就会被保留下了,如果是往不好的方面发展,就会被自然选择淘汰。 还有一种命运,就是被放置不用。复制出来以后,又加了个终止子,既不表达,也不删除, 搁那里搁着不管,成了伪基因。被保留下来的具有新功能的基因与另一个复制出来的基因之 间就是旁系同源。
异同源(Xenologs)是指通过水平基因转移,来源于共生或病毒侵染所产生的相似基因。 异同源的产生不是垂直进化而来的,也不是平行复制产生的,而是由于原核生物与真核生物 的接触,比如病毒感染,在跨度巨大的物种间跳跃转移产生的。
不同的同源,概念很容易混淆。图 1 清楚的描述了各种同源之间的关系。首先,有个早 期的球蛋白基因,它通过基因复制,形成了α球蛋白基因和β球蛋白基因。后来随着进化,这 两种复制产生的基因也存在于不同的物种中。其中某一物种里的,比如老鼠里的α球蛋白基 因和β球蛋白基因就属于旁系同源。而某一个基因在不同物种中,比如青蛙里的α球蛋白基因 和鸡里的α球蛋白基因就属于直系同源。再比如,某个细菌,它没有早期的球蛋白基因,也 自然没有β球蛋白基因,但是通过与青蛙的共生,发生了基因水平转移。于是它从某一天就 起有了β球蛋白基因。那么这个细菌的β球蛋白基因和青蛙的β球蛋白基因就属于异同源。
直系同源来自基因的分化,旁系同源来自基因的复制。
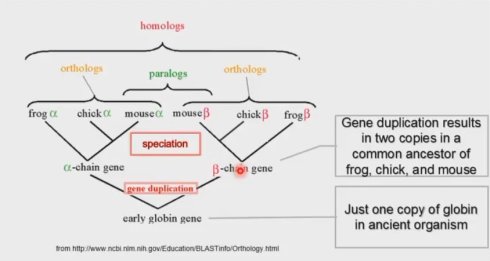
之前我们讲过两条序列的相似度如何计算,那么能不能定量描述同源呢?答案是不可以, 同源只是对性质的一种判定,只能定性描述,不能定量描述。所以“同源性等于 80%”这种 说法是错误的!
3.3 “树状” 和“网状”
越来越多的细菌和动植物的基因组测序显示,基因并不是简单遗传给生命树上的个别枝 条,它们还在物种之间以不同的进化路径转换,其结果是一个杂乱无章的“生命网”。这里 要区分“树”和“网”的概念。在计算机科学领域,树的定义规定,树上从一个点到另一个 点的路径只有唯一的一条。而当两点之间的路径个数≥2 的时候,就形成了网。编织生命网 的要素之一就是水平基因转移。水平基因转移,是指生物将遗传物质传递给其他细胞而非其 子代细胞的过程。
早在 1993 年,就有生物学家提出细菌的基因排序不是树状的,而是网状的。1999 年, 美国《科学》杂志发表言论说:“生命进化树并不是真实存在于自然界中的,而是人类用来 规划自然界的一个理论。”但是,有研究者运用更多的研究捍卫达尔文的观点,认为所谓网 状的进化论是理想化、不切实际的想法。
“树状”和“网状”的辩论在 2006 年正式拉开帷幕。位于德国海德堡的欧洲分子生物 实验室(EMBL)派出由皮尔·博克领导的工作组,研究了来自细菌、古细菌以及真核细胞 的 191 个基因组。他们发现,其中 31 组基因没有任何迹象表明曾经被水平转移过,和尚未 完善的“树”相近。但是,来自德国杜塞尔多夫大学的达冈和马汀教授认为,31 组的结果 不能够证明什么,这个数字太小。 2008 年,达冈和他的团队研究了 181 个基因组,发现 80% 的基因组存在水平基因转移,即网状树。
此外,杂交也可能是物种进化的有力驱动。来自伦敦大学的生物进化学家詹姆斯·马里 特说:“杂交是非常普遍的现象,有 1/10 的动物都是杂交的。”2008 年,美国得克萨斯大学 的科学家在包括家鼠、野鼠和非洲爪蛙在内的 8 种动物的基因组中发现了一种奇特的 DNA。 这是鸡、大象和人类所没有的 DNA,这说明它是一些动物通过异种交配形成的。几年前, 科学家也曾在牛体内发现蛇的 DNA。此外,鱼类、昆虫和植物中也都曾发现水平基因转移 现象。这些新发现意味着,有一些物种的进化是呈现树状的,而有一些却不是,所以用达尔 文的进化枝条来连接物种似乎过于简单了。
三、系统发生树介绍
3.1 系统发生树的样子
研究分子进化所要构建的系统发生树(Phylogenetic tree),也叫分子树。首先来看从系 统发生树上我们都能研究出什么?对于一个未知的基因或蛋白质序列,可以利用系统发生树 确定与其亲缘关系最近的物种。比如你得到了一个新发现的细菌的核糖体 RNA,你可以把 它跟所有已知的核糖体 RNA 放在一起,然后用他们构建一棵系统发生树。这样就可以从树 上推测出谁和这个新细菌的关系最近。系统发生树还可以预测一个新发现的基因或蛋白质的 功能。以基因为例,如果在树上与新基因关系十分密切的基因的功能已知,那么这个已知的 功能可以被延伸到这个新基因上。构建系统发生树还有助于预测一个分子功能的走势。也就 是从树上可以看出某个基因是正在走向辉煌还是在逐渐衰落。最后,系统发生树还能帮助我 们追溯一个基因的起源。甚至当它从一个物种“跳”到另一个物种上,也就是发生了水平基 因转移时,系统发生树都可以很好的展示出来。
系统发生树看上去就是很像小朋友画的简笔画(图 1)。虽然简单,但是一点也不简略, 一棵树该有的东西,它一个也没少!
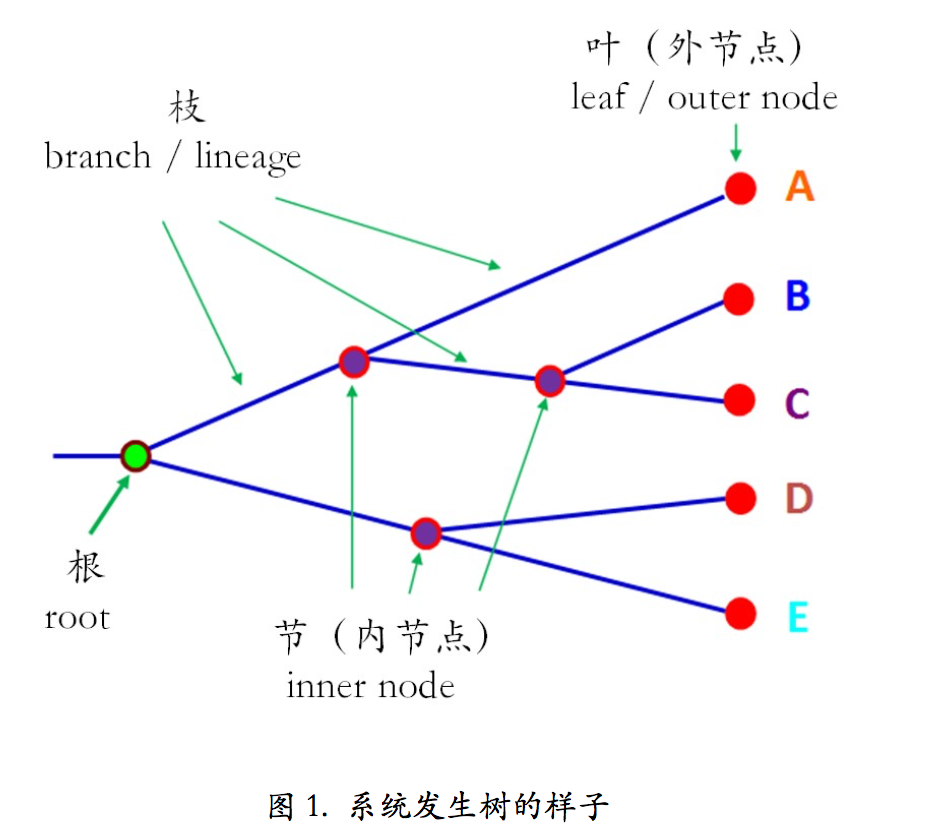
树是从根(root)长出来的。从根延伸出的树枝就叫枝(branch/lineage)。枝上有分叉, 分叉的地方就叫节(node)。枝的顶端顶着的就是叶(leaf)。根、节和叶都可以叫做节点(node)。 但是叶后面不再有枝了,是最外面的节点,所以叫外节点(outer node)。而节的前后都有枝, 所以叫内节点(inner node)。根是一切的起源,习惯上就叫根。根和节都表示理论上曾经存 在的祖先,叶子是现存的物种。这一点很重要!比如我们要研究某个基因,于是搜集了很多 物种的这个基因的序列,用它们构建了一棵系统发生树。搜集到的物种都出现在叶子上,也 就是外节点上,没有在内节点上的。内节点上都是理论上曾经存在过的共同祖先,现在已经 不存在了!
用软件创建出的系统发生树有各种形状。圆形的,方形的,三角形的等(图 2)。从生 物学意义上没有任何区别,只是表现形式不同而已。根据建树所用序列的多少来选择不同形 状的树。如果序列非常多,那么圆形的看上去就要比方的或者三角的舒服得多,便于在文献 里排版。系统发生树上从任何一个点发出的枝子围着这个点旋转都不改变树的生物学意义, 只是视觉上有点儿差别而已。所以,旋转之后的两棵树是等价的,生物学意义完全相同。

3.2 系统发生树的种类
系统发生树还分为有根树和无根树(图 1)。顾名思义,有根树就是有根,无根树就是 无根。其实两者是可以互换的。如果我们按住无根树上某一个点,然后用把梳子将树上所有 的枝条都以这个点为中心向右梳理,就能把它梳成有根树的样子。按住的这个点就是根。所 以对于一棵树来说,根的位置是主观的,你想让他在哪它就在哪里。但是你不能随意指定哪 个内节点当根,毕竟根有其自身的生物学意义,它应该是所有叶子的共同祖先。那么我们如 何确定根的位置呢?可以通过外类群(outgroup)来确定,从而把无根树变成有根树。

有根树反映了树上基因或蛋白质进化的时间顺序,通过分析有根树的树枝的长度,可以 了解不同的基因或蛋白质以什么方式和速率进化。而无根树只反映分类单元之间的距离,而 不涉及谁是谁的祖先问题。做有根树需要指定外类群。所谓外类群,就是你所研究的内容之 外的一个群。比如你要分析某一个基因在不同人种间的进化关系,那就可以额外选择黑猩猩 加入进来,作为外类群一同参与建树。或者你要分析哺乳动物,那就可以选鳄鱼、乌龟之类 的。总之,保证外类群在你要研究的内容之外,但又不能太远。外类群可以不只是一个物种, 而是多个,但也不要太多,两三个即可。为什么有了外类群之后,做出来的树就是有根树了 呢?因为你知道外类群和你研究的内容一定不是一伙的,所以外类群分支出的那个内节点就 是根。
有根树(归于一个节点)反映了树上物种或基因的时间顺序,而无根树只反映分类单元之间的距离而不涉及谁是谁的祖先问题。
1998 年,伍斯提出了一个涵盖整个生命界的系统树(图 2),也叫物种树。之后这棵树 被后人不管的补充,不断的修改,不断的完善,变得无比常庞大。物种树勾画了生物进化的 大致轮廓。从完全版的物种树上,可以找到目前人类已发现的所有有生命的东西。

我们这里讲的分子树跟物种树是有本质区别的。物种树是基于每个物种整体的进化关系, 也就是基于整个基因组构建的,而分子树是基于不同物种里某一个基因或蛋白质序列之间的 关系构建的。那么一个分子树表达出来的各物种之间的关系就可能与物种树完全不同。此时 说明这个基因经历了特殊的进化故事。也许是受到了特殊环境变化的影响,也许是发生了水 平基因转移等等。总之,这种区别的出现是很有研究价值的。
系统发生分析一般是建立在分子钟基础上的。分子钟:分子序列进化是按照一恒定速率进行的,所以积累突变的数量和进化时间成一定比例,基于这个假说,发生树上的树枝长度可以用来估算基因分离的时间。 什么叫系统进化树(Phyligenetic tree)? 系统进化树是对多序列比对(MSA)结果以树形图形式的一个呈现,对于研究进化关系有很大的帮助,通过进化树分析我们也可以关键功能基因和蛋白得出一些假说。
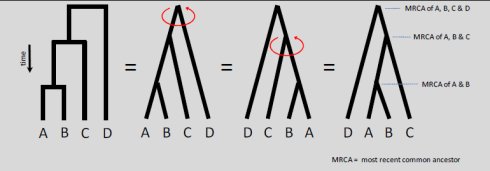
如上图所示,进化树可以有不同的表示形式
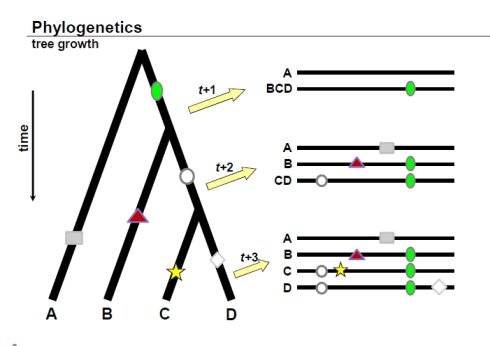
上图很好的反映了进化树构建的依据:
- 随着物种进化的演绎,进化水平越相近的物种他们的序列越接近;
- 如果是由同一个物种演化过来的,分化出来的不同物种会保留共同祖先的印记,这是区别于其他的祖先的。
四、进化树的构建
构建系统发生树的方法很多。最常用的有基于距离的构建方法,包括非加权分组平均法 (Unweighted Pair Group Method with Arithmetic mean,UPGMA),最近邻居法(Neighbor- Joining method,NJ),最小二乘法(Generalized Least Squares,GLS)等。还有最大简约法 (Maximum Parsimony,MP),最大似然法(Maximum Likelihood,ML),贝叶斯推断法(Bayesian Inference,BI)等。我们并不打算一一讲解这些方法的具体原理。你只要掌握何时选用何种 方法就可以了,算法的事自由软件替你搞定。
- 从计算速度来看,最快的是基于距离的方法, 几十条序列几秒钟即可完成。其次是最大简约法。最大似然法就要慢得多。最慢的是贝叶斯 法。
- 但是从计算准确度来看,算得最慢的贝叶斯法确是最准确,而算得最快的基于距离法结 果确是最粗糙。
- 从实用的角度,建议使用最大似然法。因为这种方法无论从速度还是准确度 都比较适中。
- 最近邻居法虽然算得快,但是当序列多,彼此差别小的时候,这种方法不适合。
- 最大简约法,似乎是个掉空里的方法,高不成低不就,所以很少有人使用。
- 贝叶斯法不是所 有的建树软件都提供,算法开发上还有待提高,而且计算时间过长。
进化树的构建大体要分为3步:序列的比对,建树,然后验证。
- 序列的比对:做ALIGNMENT的软件很多,最经常使用的有CLUSTALX和CLUSTALW
- 构建进化树有两种基本的方法:独立元素法(discrete character methods)和距离法(distance methods),基于距离的构建方法UPGMA(Unweighted pair group method with arithmetic mean,平均连接聚类法)、ME(Minimum Evolution,最小进化法)和NJ(Neighbor-Joining,邻接法);基于特征的构建方法:最大简约法(MP法),最大似然法(ML法),进化简约法(EP法),相容性方法等。
不同的方法可能会得到不同的结论,我们需要用不同的方法以及不同的参数,加上对生物问题的理解来构建最好的进化树来帮助我们更好的理解生物学问题。其中一个衡量树的好坏的方法就是看bootstrap的值,值越大越好,
距离法:
- UPGMA (unweighted pair group method with arithmetic means)
- Fitch-Margoliash
- Neighbor-joining
距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的。进化树枝条的长度代表着进化距离
1,所有的距离法首先通过俩俩比对产生一个“距离矩阵”,然后计算出每对序列的基于距离,简单的理解基因距离就是两个序列没有匹配上的个数(当然,实际计算比这个要麻烦的多);
2,然后这个俩俩比对距离矩阵用来判断距离最近的两个序列,这两个序列来形成进化树的两个树枝,这些俩俩比对的距离矩阵然后重新开始找序列最近的两个序列,但这次最相近的两个序列通过一个节点连接到树上,以次往下推,直到结束。
3,再根据距离画好这个树。 这个方法的优点就在于快速,缺点为:
- 它的准确与否是建立在这样的假说上的:additive distances (always)和molecular clock (sometimes);
- Information loss occurs due to data transformation ;
- Uninterpretable branch lengths;
- Single “best tree” found.
独立元素法:
- Parsimony
- Maximum likelihood
所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的(例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了)。根据transition probabilities, base frequencies, rate heterogeneity等等求出最大的概率图
举个硬币的例子来说明问题吧
Likelihood (L) = Probability (dataobserved | model)
Data : HHTHTH
Model 1 : fair coin Prob(H) = 0.5, Prob(T) = 0.5
Model 2 : 2-head coin Prob(H) = 1.0, Prob(T) = 0.0
Model 3 : 2-tail coin Prob(H) = 0.0, Prob(T) = 1.0
L (Data|Model1)
= Prob(H|Model1) * Prob(H|Model1) * Prob(T|Model1) * Prob(H|Model1) *
Prob(T|Model1) * Prob(H|Model1)
= 0.5 * 0.5 * 0.5 * 0.5 * 0.5 * 0.5 = 0.0156
L (Data|Model2) = 1.0 * 1.0 * 0.0 * 1.0 * 0.0 * 1.0 = 0.0
L (Data|Model3) = 0.0 * 0.0 * 1.0 * 0.0 * 1.0 * 0.0 = 0.0
同理对于maximum likelihood
Find the model that maximizes the likelihood of the observed data
Data : GGACGCCTGACGCCGCTCGG
Model 1: equal base composition - 0.25, 0.25, 0.25, 0.25 – A, C, G, T, respectively
Model 2: G+C bias - 0.1, 0.4, 0.4, 0.1 – A, C, G, T, respectively
Model 3: A+T bias - 0.4, 0.1, 0.1, 0.4 – A, C, G, T, respectively
L (Data|Model1) = Prob(G|Model1)*Prob(G|Model1)*Prob(A|Model1)*...* Prob(G|Model1) = 0.2520 = 9.1x10-13
L (Data|Model2) = 0.416 * 0.14 = 4.3x10-11 ← maximum likelihood
L (Data|Model3) = 0.116 * 0.44 = 2.6x10-18
对于核酸替代进化模型需要考虑两个因素:1每个碱基出现的概率;2根据进化关系确定的每个碱基转移的概率。有很多进化模型,合理选择。
优点:基于精细的进化模型;可以对特殊的树形拓扑图的相似性统计评估;经常返回很多相似的图(Often returns many equally likely trees),比其他的方法呈现的结果要好。
缺点:计算花时间,Often returns many equally likely trees.
3,Bootstrap验证
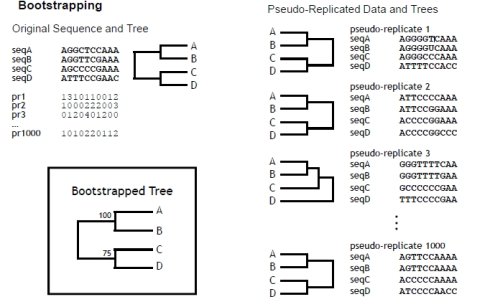
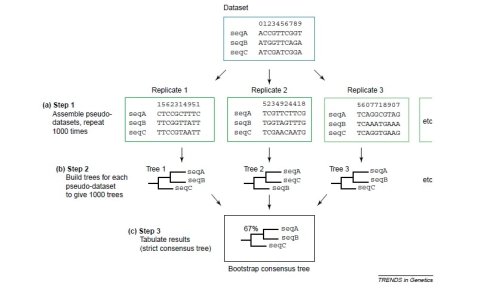
这是目前公认的比较好的检验方法,采用随机抽样的方法组成新的序列,然后序列比对,出现同样的比对结果的概率,如下图所示,我们对一定长度序列有放回的抽取,抽取次数跟序列长度一样,pr1为第一次随机抽样的模式,第一个碱基被抽中了1次,第二个碱基被抽中了3次,第三个碱基被抽中了1次……然后以这种模式抽提所有的比对的序列形式新的序列,然后在对新的序列进行构图,同理,随机产生新的模式,重复之前的步骤,这样的随机产生的模式一共1000组,最后求出模中节点出现的概率即为该序列比对的bootstrap概率。
这种方法需要两种假设:数据量足够大;The data are identically and independently distributed。
Bootstrap values
> 90% strongly supported
70 > 90% well supported
50 > 70% weakly supported
< 50% not supported
上面两个图bootstrap原理一样,但是在建立取点模式上有一点点不一样。
五、对进化树的分析
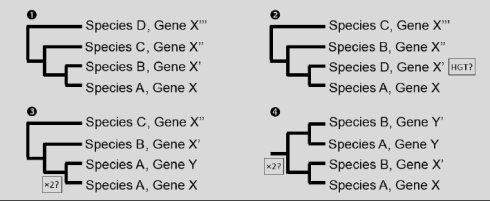
假设上面的比对结果都有一个不错的bootstrap验证,基因X’,X’’,X’’’,X’’’’直系同源,Y,Y’跟这几个旁系同源(直系来源于基因的分化,旁系来源于基因的复制),图1是我们的参考进化图,反应的跟我们预期的一样,图2中跟我们预期的图1一比,我们怀疑可能是发生了基因水平转移(HGT);图3,我们发现了一个旁系同源基因,在其他的物种中没有发现旁系同源,所以我们怀疑可能是发生了基因复制;图4我们看到旁系同源基因也跟其他的物种同源,可以推断出这个复制的过程在这两个物种的祖先就已经发生了。
六、常用工具
推荐这个快速做进化树的在线工具:
目前流行的建树软件(表 1),比如 PHILIP 和 MEGA,基本能够包括上述所有算法。 如果想要构建 ML 树,也可以尝试专门构建 ML 树的 PHYML。贝叶斯的算法以 MrBayes 为 代表,只是计算速度比较慢。如果构建的系统发生树要用于发表生物信息学领域的文章,需 要两种以上的构建方法锁定同一个结果才能审稿通过。如果是用于发表以生物实验为主的文 章用一种构建方法就可以了。
表1 构建分子进化树相关的软件
| 软件 | 网址 | 说明 |
|---|---|---|
| ClustalX | http://bips.u-strasbg.fr/fr/Documentation/ClustalX/ | 图形化的多序列比对工具 |
| ClustalW | http://www.cf.ac.uk/biosi/resear … loads/clustalw.html | 命令行格式的多序列比对工具 |
| GeneDoc | http://www.psc.edu/biomed/genedoc/ | 多序列比对结果的美化工具(可以导入fasta格式的文件,出来的图可用于发表,我用过) |
| BioEdit | http://www.mbio.ncsu.edu/BioEdit/bioedit.html | 序列分析的综合工具 |
| MEGA | http://www.megasoftware.net/ | 图形化、集成的进化分析工具,不包括ML |
| PAUP | http://paup.csit.fsu.edu/ | 商业软件,集成的进化分析工具 |
| PHYLIP | http://evolution.genetics.washington.edu/phylip.html | 免费的、集成的进化分析工具 |
| PHYML | http://atgc.lirmm.fr/phyml/ | 最快的ML建树工具 |
| PAML | http://abacus.gene.ucl.ac.uk/software/paml.html | ML建树工具 |
| Tree-puzzle | http://www.tree-puzzle.de/ | 较快的ML建树工具 |
| MrBayes | http://mrbayes.csit.fsu.edu/ | 基于贝叶斯方法的建树工具 |
| MAC5 | http://www.agapow.net/software/mac5/ | 基于贝叶斯方法的建树工具 |
| TreeView | http://taxonomy.zoology.gla.ac.uk/rod/treeview.html | |
| 进化树显示工具(加红色标注的为最通用的分析软件) |
ps:个人感觉这个的整理非常强大,好多东西都搞明白了
参考资料
- 柳城的博客:http://liucheng.name/577/
- BIOINFOMATIC METHOD课程
- Baldauf S L. Phylogeny for the faint of heart: a tutorial[J]. TRENDS in Genetics, 2003, 19(6): 345-351.
- 山东大学 生物信息学课题组荣誉出品 http://www.crc.sdu.edu.cn/bioinfo 巩晶老师课件
