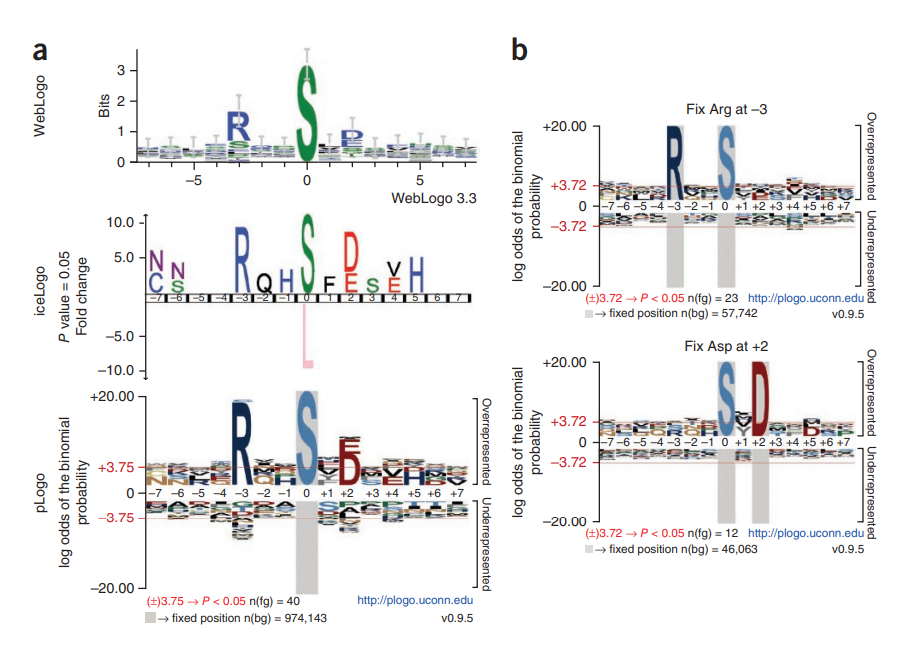
【9.1.5】序列标识图-plogo
用于可视化蛋白质或核酸基序的方法传统上依赖于残基频率以图形方式缩放字符高度。我们描述了 pLogo,这是一种图案可视化,其中残基高度相对于其统计显着性进行缩放
二、原理介绍
用于 pLogo 生成的位置权重矩阵 (PWM) 分数。
pLogo 是 PWM 的可视化表示,其中每个可能的残基在每个位置都有一个值。这些值基于给定背景数据集中该残基的概率的前景数据集中该残基的频率的统计显着性。因此,一个 15 个碱基对的 DNA 基序总共有 15 个位置 × 4 个核苷酸 = 60 个值,一个 15 个残基的蛋白质基序将有 15 个位置 × 20 个氨基酸 = 300 个值。在 pLogo 中,这些值中的每一个都由特定残基在x轴上方或下方的高度可视化表示,具体取决于值的符号(+ 或 -)。如下所述计算每个位置的每个残基值。
为了确定单尾显着性,人们通常会计算概率分布所需一侧下的面积。使用这种方法,需要使用逻辑和案例来确定要整合的分布的适当一侧(即,代表性过高与代表性不足)。然而,对于 pLogo 生成,需要一个连续的基于概率的分数,跨越代表性不足、过度代表性和介于两者之间的值。因此,我们选择使 pLogo 中的残基高度与过度代表性的重要性与代表性不足的重要性的对数几率密切相关。这些显着性值是使用残基频率的二项式概率计算的,相对于基因组或蛋白质组背景,使用以下公式:
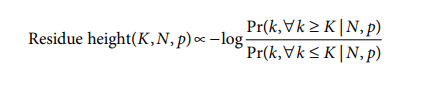
其中K是给定位置特定类型残基的实际数量,N是某个位置的残基总数,p是该位置出现残基的概率。p的值来自背景总体(参见“背景概率的计算”),使得
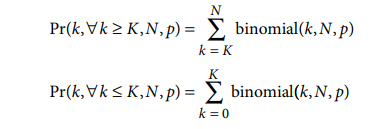
请注意,在此公式中,恰好K次出现的概率Pr( K , N , p ) 实际上是重复计算的,因为它包含在分布的两个尾部中。因此,因为两个尾部的总和将略大于 1.0,所以这不是严格的对数赔率,而是非常接近的近似值。此外,随着N 的增加,残留高度接近精确的对数优势值,因为 Pr( K , N , p ) 向 0 减小。在⌊ N × p ⌋ ≤ K ≤ ⌈ N × p⌉ 的情况下,这个对数几率近似值将接近 0,这是当残基既不代表不足也不代表过多时的期望结果。
使用 pLogo 残基高度的二项式概率的这种对数几率近似具有以下吸引人的特性:(ii) 当残基既不代表不足也不代表过高时,它的值为 0(或非常接近于 0);(iii) 对于从 0 ≤ K ≤ N 的每个K值,它都有明确的定义;(iv) 它是单调的并且随着K接近N × p的值而接近 0 ;(v) 它快速逼近真实对数赔率以及 –log(Pr( k , ∀ k ≥ K | N , p )) 或 –log(Pr( k , ∀ k ≤ K | N , p )) 因为K离N × p更远。(因此,如果一个残基的代表性严重不足或过高,一个概率将非常小,另一个将接近 1.0;因此,朝向一条尾巴或另一条尾巴几乎不会受到另一条尾巴权重的影响)。
所有log均以 10 为基数计算。
如果提供参考基因组,概率p被计算为背景中那个位置的残基的分数。
显著性统计
红色水平 pLogo 条在比较多个 pLogo 时提供了一个有用的参考点,它允许快速确定特定基序位置中最具统计意义的过度或代表性不足的残基是否超过所选的α值(通常为 0.05)。为了准确地确定条的位置,必须纠正在生成 pLogo 时进行的统计测试的数量。在 pLogo 生成的任何给定迭代中执行的统计测试数量的确定等价于计算的二项式概率的数量,可以通过以下公式确定:
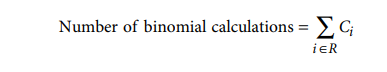
其中R表示具有非固定残基的位置集合,C i表示在位置i的背景集合中唯一字符的数量。通常,对于 DNA 或 RNA 等核酸序列,C i 的所有值都等于 4,对于蛋白质而言,C i 的所有值都等于 20,但情况并非如此,上述公式在本质上是通用的。因此,对于预期的α值为 0.05 ,使用保守的 Bonferroni 校正,可以使用以下公式在对数刻度上放置统计显着性条

请注意,统计显着性条的位置取决于 pLogo 中固定位置和独特字符的数量。
参考资料
- 2013,pLogo: a probabilistic approach to visualizing sequence motif 。 https://www.nature.com/articles/nmeth.2646
