【4.9.1】基于bam文件质量评估工具 qualimap
一、qualimap简介
qualimap可用于统计bam文件,输出结果包含可视化图形和详细统计信息,以及每条contig的mapping信息。
软件官网及案例:
http://qualimap.conesalab.org/ 案例 http://qualimap.conesalab.org/doc_html/samples.html#bam-samples
二、安装
wget https://bitbucket.org/kokonech/qualimap/downloads/qualimap_v2.2.1.zip
# 解压
unzip qualimap_v2.2.1.zip
# 进入软件目录
cd qualimap_v2.2.1
# 查看帮助信息
./qualimap -h
软件模块:
bamqc: 用于单个bam文件的QC统计
rnaseq: 用于转录组RNA-Seq样本的bam文件的QC统计
multi-bamqc: 用于多样本的bam文件分组QC统计
counts: 用于转录组数据计数的统计,用于量化表达水平
clustering: 用于表观遗传特征的聚类
comp-couts: 输入bam文件和注释文件,计算映射到每个区域reads的数量
三、运行
3.1. 执行bamqc模块命令
bamqc模块用于单个NGS样本bam文件的统计。
qualimap bamqc -bam sample.bam \ # 指定bam文件路径
-outformat PDF:HTML \ # 输出文件格式
-outdir out \ #输出文件目录
-nt 12 \ # 线程数
--java-mem-size=10G #设置最大内存
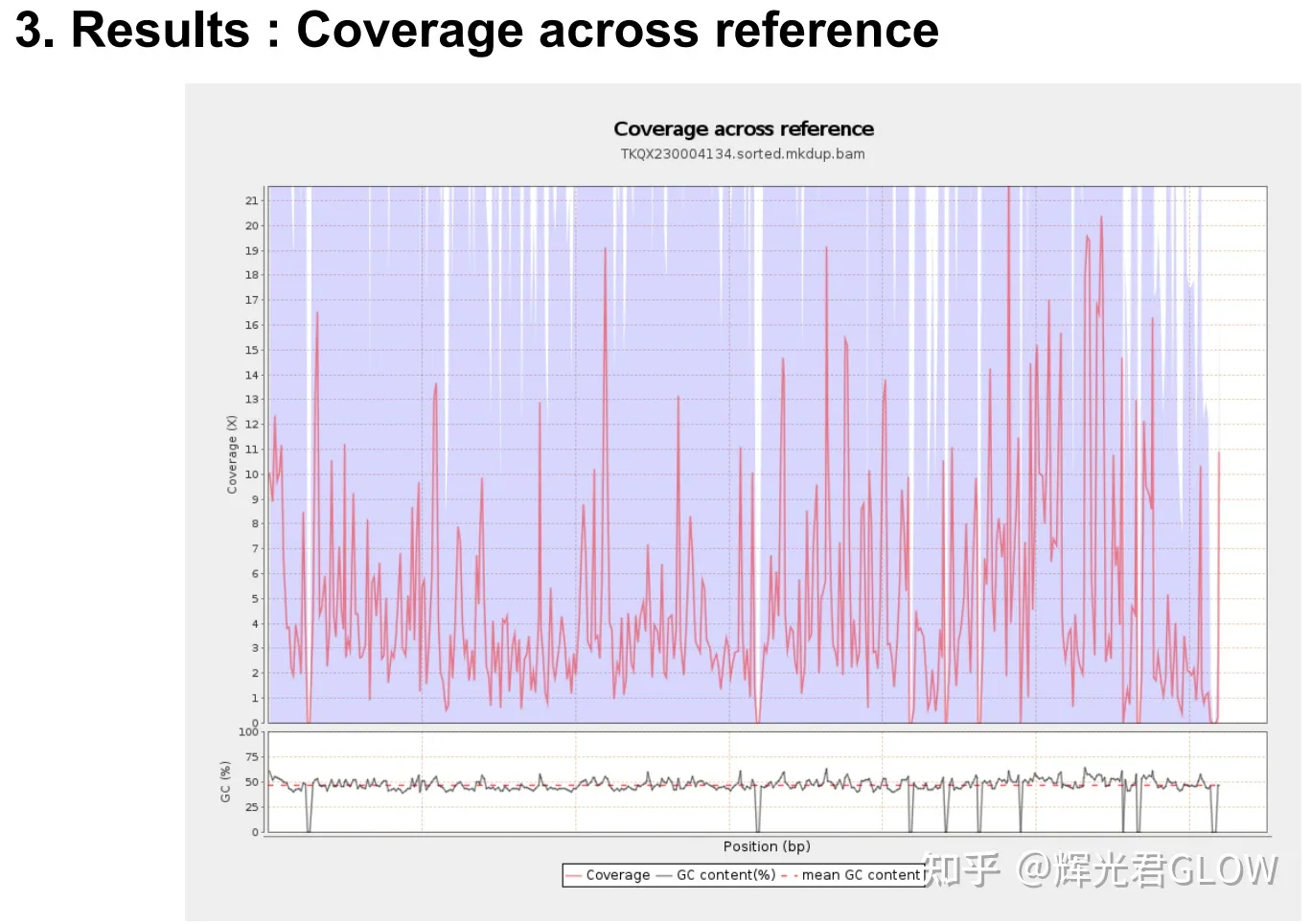
程序运行结束后,统计信息在report.pdf文件中查看。
report.pdf中部分图表如下所示:
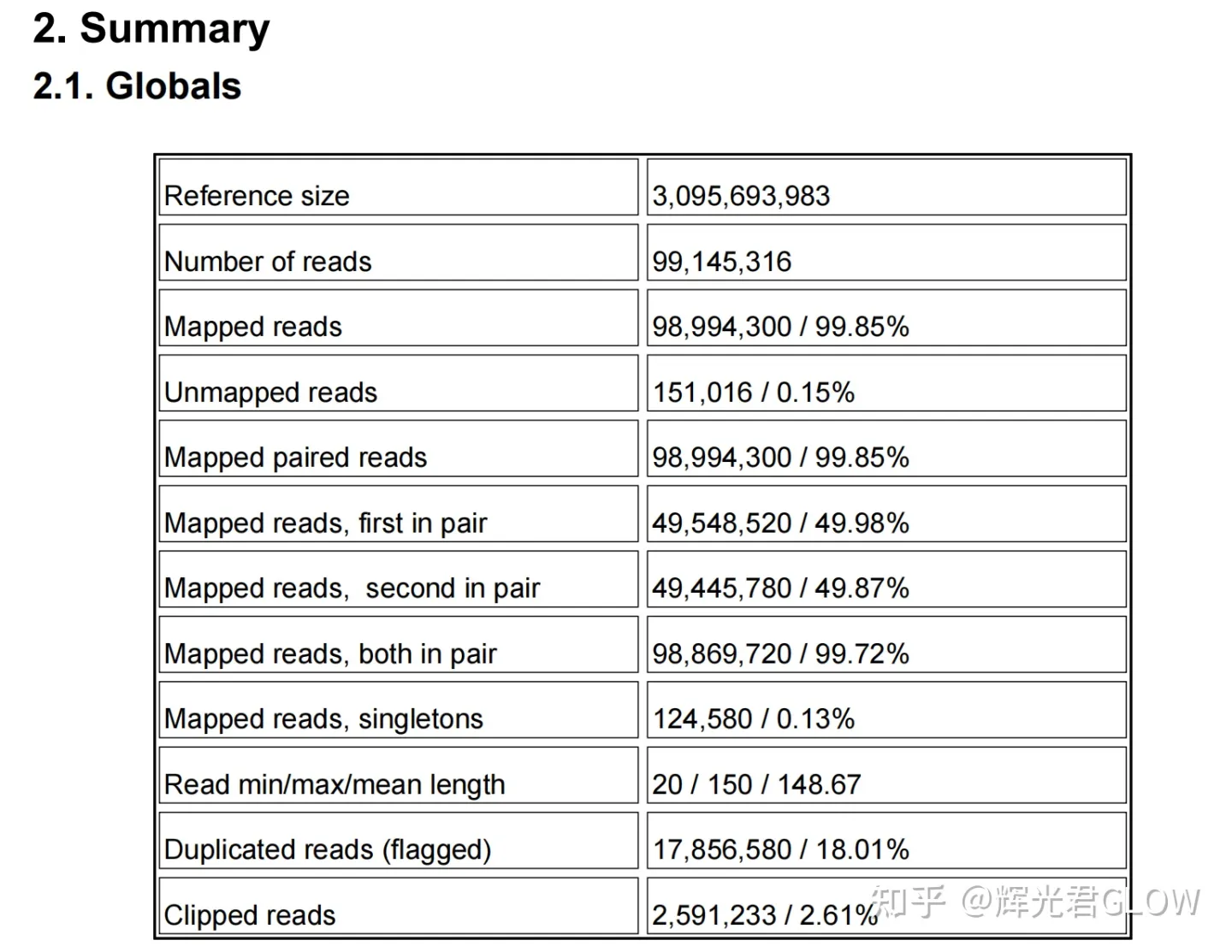
Summary - Globals
3.2. 执行rnaseq模块命令
rnaseq模块用于RNA-seq数据的bam文件的统计。
qualimap rnaseq -bam sample.bam \ # 指定bam文件路径
-outformat PDF:HTML \ # 输出文件格式
-outdir out \ #输出文件目录
-nt 12 \ # 线程数
--java-mem-size=10G #设置最大内存
3.3. 执行multi-bamqc模块命令
multi-bamqc模块用于多样本NGS的bam文件的统计和比较。
qualimap multi-bamqc -r \ # -r指定输入bam文件
-d qualimap.list \ # 输入文件列表
-outformat PDF:HTML \ # 输出文件格式
-outdir out \ #输出文件目录
-nt 12 \ # 线程数
--java-mem-size=10G #设置最大内存
其中输入文件列表qualimap.list有三列,每行一个样本,第一列样品名称,第二列 包含路径的bam文件/bamqc结果目录,第三列组名。
四、我的案例
qualimap bamqc -bam ${output}/${ID}/${ID}.bam -outdir ${output}/${ID}
参考资料
这里是一个广告位,,感兴趣的都可以发邮件聊聊:tiehan@sina.cn
![]() 个人公众号,比较懒,很少更新,可以在上面提问题,如果回复不及时,可发邮件给我: tiehan@sina.cn
个人公众号,比较懒,很少更新,可以在上面提问题,如果回复不及时,可发邮件给我: tiehan@sina.cn
