【9.1.4】寻找保守序列-PRINTS指纹图谱数据库
目前,科学家已经对现有的蛋白质序列进行了充分的研究,而且早已发现并总结了这些 序列上的重要基序。相关研究成果汇入了 PRINTS 蛋白质序列指纹图谱数据库( http://www.bioinf.manchester.ac.uk/dbbrowser/PRINTS/ )。所谓蛋白质的指纹是指一组保守的序列基序, 用于刻画蛋白质家族的特征。这些基序由多序列比对结果获得,且它们在氨基酸序列水平上 是不相邻的,但是在三维结构中可能紧密地结合在一起。PRINTS 数据库存储了目前已发现 的绝大多数蛋白质家族的指纹图谱。对于一个陌生的蛋白质,只要看看它的序列是否符合某 个蛋白质家族的图谱就可以对它进行分类并预测它的功能。
要浏览 PRINTS 数据库,可以输入数据库编号、关键词、或标题等以查找某一个指纹图 谱。比如点击“By text”通过关键词搜索(图 1)。输入条中输入“TRANSFERRIN”,也就 是搜索转铁蛋白家族的图谱。搜索返回转铁蛋白家族的指纹图谱链接。

点击结果页面中的“TRANSFERRIN”链接后,会显示包括指纹图谱的基本信息、与其 他数据库之间的交叉链接、构建指纹图谱所使用的蛋白质序列、以及指纹图谱中每个基序等具体信息(图 2)。

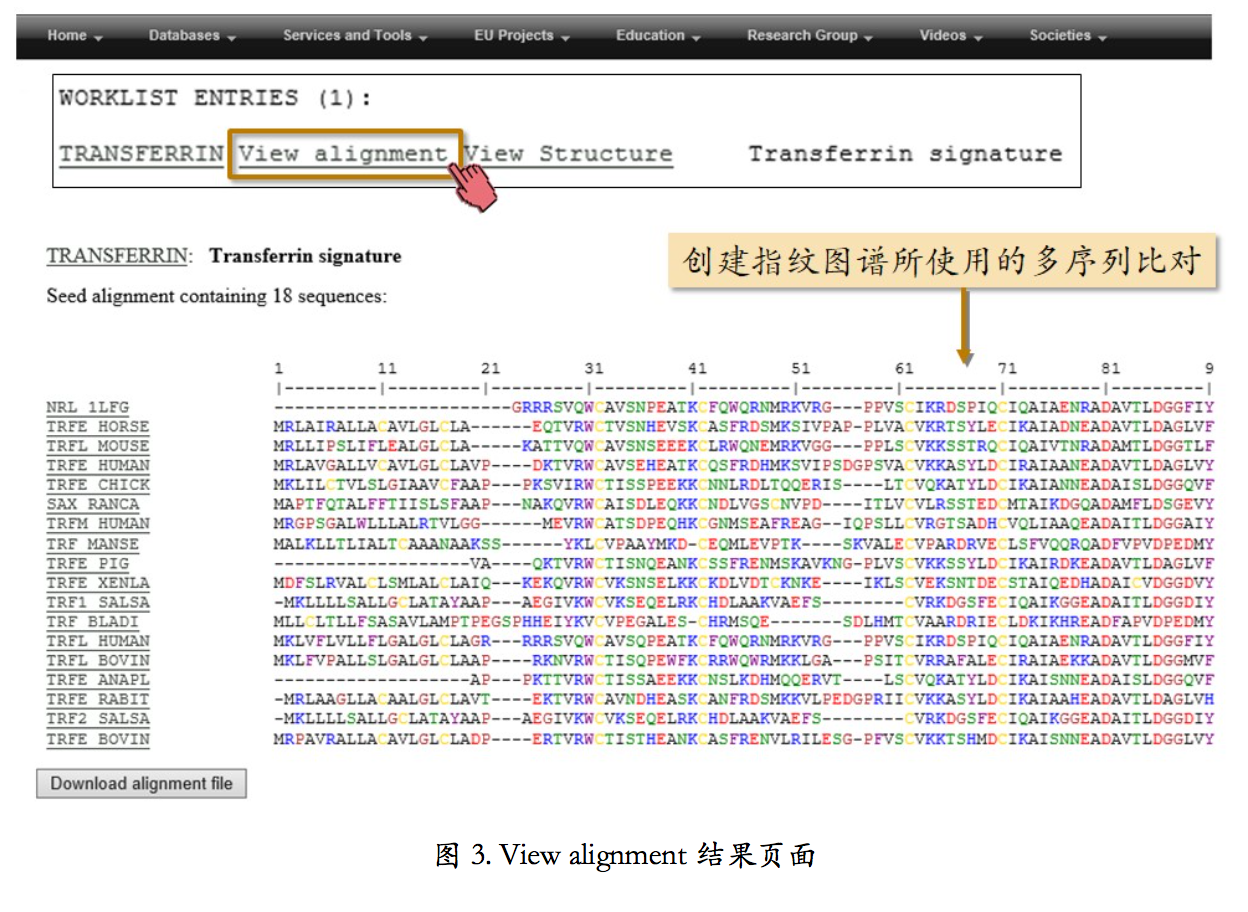
点击“View alignment”链接后,可以看到创建指纹图谱所使用的多序列比对(图 3)。
点击“View structure”链接后,网页会打开一个三维视图插件,并以该家族中某一特征 蛋白质具有的三维结构为例,在线显示指纹图谱中各个基序在三维结构中的位置(图 4)。 从该三维结构图中可以看出,紫色的基序在氨基酸序列水平上并不相邻,但是在三维空间结 构中是紧密联系在一起的,并形成蛋白质的重要功能区。

除了浏览某一指纹图谱,PRINTS 还提供指纹匹配服务。也就是搜索某一序列所匹配的 指纹图谱。此功能通过 PRINTS 主页也上的“FPScan”链接实现(图 5)。注意输入的待搜 索序列只能是“a raw sequence”,也就是纯序列。换言之,FASTA 格式中带大于号的第一行 不能拷贝进输入框。示例文件 prints.fasta 请从课程附件中下载。
提交后返回的结果页面中,跟输入序列匹配的指纹图谱,根据匹配得分的高低被排列出 来(只列出前十名)(图 6)。此外,还单独列出了排名前三的指纹图谱。由此可知,得分最 高的是视紫红质家族的指纹图谱。


点击排名第一的视紫红质家族的“Graphic”链接,可以得到该家族指纹图谱中各个基 序在输入序列中所匹配的位置(图 7)。结果页面的下部还提供了视紫红质家族的 6 个基序 在输入序列中所对应的具体序列片段。由此,可以推测,输入序列属于视紫红质家族,并具 有该家族蛋白质的主要功能。事实上,输入序列确实是从 UniprotKB 数据库中下载的一条羊 的视紫红质的序列(P02700)。

二、其他

PRINTS数据库是蛋白质基序指纹的纲要。每个指纹都已在ADSP或VISTAS序列分析软件包中使用数据库扫描过程进行了定义和迭代完善。数据库中代表两种类型的指纹,即根据其复杂性,它们可以是简单的也可以是复合的。复合指纹则编码多个motifs。数据库条目的大部分属于后一种类型,因为多分量搜索的判别能力更大,因此结果更易于解释。
编译此类数据库有两个主要原因:
- 合理化OWL复合序列数据库中包含的大量数据(即将序列分解为家族,超家族和亚家族);
- 因此,将来可以提高序列分析的效率。
有两种直接的方法可以使序列分析更有效:
- 首先,可以对整个数据库运行新序列,以获取有关结构或功能的可能线索;
- 其次,可以提取数据库条目以针对单个序列或个人数据库运行。
两种选择本质上都是基于知识的,因此与整个复合数据库的耗时搜索相比,可以非常快速地进行诊断。
1.1 motif什么意思?
作为序列比对的任何保守元素的基序:是与功能或结构已知或其重要性可能未知的区域相对应的局部比对。 它是保守的就足够了,因此很可能预示了任何其他蛋白质序列中这种结构/功能区的任何后续出现。
1.2 fingerprint什么意思?
指纹是一组motifs,用于预测单个序列或数据库中相似motif的出现。通过迭代扫描OWL复合序列数据库来完善指纹,即增强其诊断能力。具有这样排列的基序的数据库搜索本质上是频率扫描:即,在其最基本的应用中,没有二级结构信息,相似性数据或任何描述的加权方案用于提高区分能力。
复合或多基序指纹包含从多重比对的不同部分获取的多个比对的motif。在这些系统中,由于对指纹各个元素的识别是互为条件的,因此增强了识别能力。这样,真正的家庭成员就容易拥有指纹的所有元素,而仅通过拥有部分指纹就可以识别出真正的家庭成员。
1.3 指纹如何生成?
指纹识别方法依赖于以下事实:在任何蛋白质家族中,序列的仅一部分是共有的–这些通常与关键功能区域或折叠的核心结构元件有关。 因此,指纹定义的起点是多序列比对,这是我们使用SOMAP,XALIGN或VISTAS手动比对程序实现的。 初始对齐只需要包含少量数字,因为该方法本身旨在随每次数据库扫描添加到对齐中。 一旦识别出一个或一组图案,就以局部比对的形式切除保守区。 关于这些图案的并置没有任何规则,除了它们基本不应该重叠:这样,图案可以彼此相邻出现,或者可以沿着排列的长度分开任何距离。
对每个对齐的基元进行独立的数据库扫描,从而生成一组命中列表,每个基元一个。然后对命中列表进行分析或关联,以确定数据库中哪些序列与指纹的所有元素匹配,哪些仅与指纹的一部分匹配。只有那些与所有元素匹配的序列才被视为真实匹配。如果搜索效果良好,则真实集合将包含比原始比对更多的序列。然后,使用来自新真实集合的其他序列数据生成另一组比对的基序,并再次搜索数据库-被诊断的家族非常大,在下次扫描之前从比对中删除多余的基序。重复此过程,直到收敛为止,在该点上,真实集在连续扫描之间保持恒定。
该迭代过程中最终对准的图案构成了精致的指纹,该指纹已输入到PRINTS数据库中。好的指纹会发现OWL中存在的所有真实匹配项,它们显示出清晰的区分界限,并且几乎没有噪声。有时,临界值可能难以评估:这通常是亚家族鉴定的结果,其中识别出仅以指纹的一部分为特征的序列子集;但也可能是由于在指纹中仅使用2个或3个图案-识别力随所用图案的数量而提高。
参考资料
- 山东大学 生物信息学课题组荣誉出品 http://www.crc.sdu.edu.cn/bioinfo 巩晶老师课件
- http://130.88.97.239/PRINTS/printsman.php
- https://www.ebi.ac.uk/training/online/course/introduction-protein-classification-ebi/what-are-protein-signatures/signature-types/what-ar-0
