【7.4.5】高度可解释的神经编码器-解码器网络从肠道微生物组预测肠道代谢物
-
新一代测序 (NGS) 和色谱分析 [例如液相色谱质谱 (LC-MS)] 的技术进步使鉴定数千种微生物和代谢物种类并测量它们的相对丰度成为可能。在本文中,我们提出了一个稀疏神经编码器-解码器网络,以根据微生物丰度预测代谢物丰度。
-
使用来自一组炎症性肠病 (IBD) 患者的配对数据,我们表明我们的神经编码器-解码器模型在准确性、稀疏性和稳定性方面优于线性单变量和多变量方法。重要的是,我们表明我们的神经编码器-解码器模型不仅仅是一个旨在最大化预测准确性的黑匣子。相反,网络的隐藏层(即潜在空间,仅由稀疏加权的微生物计数组成)实际上捕获了本身具有临床意义的关键微生物-代谢物关系。虽然这个隐藏层是在不了解患者诊断的情况下学习的,但我们表明,学习到的潜在特征的结构方式可以高精度地预测 IBD 和治疗状态。
-
通过施加非负权重约束,网络成为一个有向图,其中每个下游节点都可以解释为上游节点的加法组合。在这里,中间层包含不同的微生物-代谢物轴,这些轴将关键微生物生物标志物与代谢物生物标志物联系起来。通过使用组成数据分析方法预处理微生物组和代谢组数据,我们确保我们提出的多组学工作流程将推广到任何一对组学数据。据我们所知,这项工作是神经编码器 - 解码器在多组学生物数据的可解释整合中的首次应用。
一、背景
人类肠道是一个复杂的生态系统,宿主细胞和外来生物在其中共存、合作和竞争。悬浮在这个生态系统中的是营养代谢物的环境,就像货币一样,被生活在环境中的生物体交换和转化。新一代测序 (NGS) 和色谱分析 [例如液相色谱质谱 (LC-MS)] 的技术进步使鉴定数千种微生物和代谢物种类并测量它们的相对丰度成为可能。通过对粪便样本应用 NGS 和 LC-MS,人们对肠道细菌产生、消耗和诱导代谢环境的复杂生态系统获得了两种互补的“观点”。这些数据模式都加深了我们对炎症性肠病等难以捉摸的肠道疾病的理解。1 ],并且越来越多地并行收集 [ 2 – 4 ]。
曾经很少遇到的炎症性肠病 (inflammatory bowel disease,IBD) 已成为发达国家的主要健康负担,自第二次世界大战以来其发病率稳步上升 [ 5 ]。IBD 是克罗恩病 (Crohn’s disease,CD) 和溃疡性结肠炎 (UC,ulcerative colitis) 这两种不同临床综合征的总称,两者的特征都是由遗传和环境因素引起的胃肠 (GI) 道慢性免疫紊乱 [ 6 , 7] ]。虽然 CD 表现为胃肠道任何部分的斑片状透壁(深部)炎症,但 UC 的特征是从直肠延伸到结肠的弥漫性粘膜(浅表)炎症 [ 8]]。尽管 IBD 的炎症没有感染源,但 CD 和 UC 患者表现出不规则的肠道微生物组,细菌多样性较少,健康细菌耗尽,不健康细菌过多 [ 5 , 9 , 10 ]。这些变化部分归因于对良性共生生物的异常免疫反应 [ 5 ]。称为生态失调的微生物组不规则性也与微生物功能 [ 11 ] 和代谢特征 [ 12 ] 的同时变化有关,这些变化共同会破坏正常的肠道生理机能。例如,Marchesi 等人。在 IBD 粪便样本中发现低水平的短链脂肪酸 [ 13],这可能与肠道细菌代谢碳水化合物的方式发生变化有关 [ 1 ]。
弗朗索萨等人研究了 164 名 IBD 患者和 56 名健康对照的配对微生物和代谢特征,产生了同类中最大的公开可用多组学数据集之一 [ 14 ]。在他们的多组学分析中,作者报告说,所有可能的成对关联中只有 6% 具有统计学意义,得出的结论是代谢物“倾向于不机械地关联”与微生物组 [ 14 ]。然而,多组学数据集成可以通过多种方式进行,从简单到复杂。我们将这些方法分为四层。
- 第一个,也是最简单的,使用迭代单变量-单变量回归,例如,测量单个细菌和单个代谢物之间的 Pearson 相关性(如 Franzosa 等人所做的那样。[ 14]])。这种简单的方法在几个特定于微生物组的软件工具中实现了 [ 15 ]。尽管成对关联很容易解释,但它们缺乏对细菌(或代谢物)共现的累加效应进行建模的能力。
- 第二种使用迭代单变量-多变量回归,例如,测量单个细菌作为所有代谢物的函数(反之亦然)。这种方法仍然易于解释,并且已被应用于从 DNA 突变推断基因表达 [ 16 ],以及来自微生物组的代谢物变量 [ 2]]。
- 第三种使用单一的多元-多元回归,例如典型相关 (CanCor) 分析。CanCor 是一种强大的工具,可以找到与代谢物组合最大程度相关的微生物组合。该技术先前已应用于研究 IBD 患者中挥发性呼吸代谢物与肠道微生物组之间的关系 [ 17 ],但其广泛应用受到高维 [ 1 ](至少没有正则化 [ 18 ])的限制。
- 为了进一步深入建模,我们探索了第四层,它利用多层神经网络对单个多元-多元回归进行建模。这些网络的隐藏层就像一个交换机,通过一组可以学习层之间复杂(非线性)模式的中间节点将输入层与输出层连接起来。在生物学中,深度神经网络已被用于许多应用,包括从 1,000 个标志性基因中预测 20,000 个基因的表达 [ 19 ]。虽然有用,但这些网络的隐藏层没有将模型连接到真实生物过程的自然意义。为了解决这个限制,许多神经网络遵循编码器 - 解码器模式,其中网络具有沙漏形状,具有压缩输入输出关系的狭窄中间层。该层被正则化为低维,因此它只有足够的信息空间来描述输入-输出转换。因此,过滤掉所有其他信息。这一层将网络分为两个专门的部分:编码器和解码器。人们可以将通用编码器-解码器网络视为神经“信号转换器”,旨在通过中间表示Z将一个数据集X转换为另一个数据集Y。
编码器-解码器网络已在计算机视觉中以完全卷积模型 [ 20 ]的形式进行了研究,其中图像被编码为紧凑的表示,然后被解码为所需的特征图。在生物医学成像中,U-net [ 21 ] 已经成功地使用编码器和解码器分割细胞图像,形成两个相互作用的 U 形轴。在生物学中,自动编码器——一种特殊类型的编码器-解码器架构,其中输入和输出是相同的——已被用于对酵母、假单胞菌和癌细胞进行聚类,其中隐藏层据称提供了具有生物学意义的数据抽象[ 22]]。然而,将一个数据域转换为另一个数据域的通用编码器-解码器显然尚未用于多组学数据集成。在这种通用形式中,编码器-解码器可以像 CanCor 一样工作,通过潜在空间预测来自多个输入的多个输出。与 CanCor 不同,编码器-解码器学习特征之间的深层非线性关系
使用编码器-解码器架构,我们试图找到一个好的模型,该模型可以在多组学数据中提供简单、描述性和可验证的模式。在这份手稿中,我们介绍了我们高度可解释的神经编码器-解码器,旨在了解肠道微生物组与其周围代谢物之间的非线性和协同关系。通过这样做,我们证明了
- 神经网络在微生物组-代谢组预测方面优于线性模型,
- 网络稀疏化以及非负权重约束提高了编码器的准确性、稳定性和可解释性 -解码器模型。
重要的是,我们表明我们的神经编码器-解码器模型不仅仅是一个旨在最大化预测准确性的黑匣子。相反,网络的隐藏层(即潜在空间,仅由稀疏加权的微生物计数组成)实际上捕获了本身具有临床意义的关键微生物-代谢物关系。虽然这个隐藏层是在不了解患者诊断的情况下学习的,但我们表明,学习到的潜在特征的结构方式可以高精度地预测 IBD 和治疗状态。总之,我们的工作表明,可以使用神经网络集成成对的多组学数据,该网络的隐藏层在没有任何监督的情况下抽象出具有临床意义的输入数据表示。通过使用标准组合方法预处理数据,我们确保我们的编码器-解码器工作流程将推广到任何一对组学数据。
二、方法
2.1 数据采集和预处理
文章从弗兰佐萨等人的补充中获得了成对微生物群和代谢体数据作为原始数据。为了降低数据的维度,实验删除了超过 50% 的测量值为零的特征,并且用cmultRepl软件的(cmultRepl零替换函数)零替换功能将剩余的零替换成非常小的数字。
2.2 数据处理
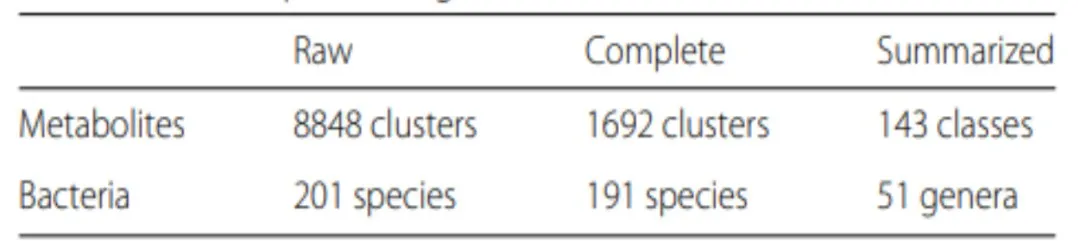
进行两个操作处理数据:“complete"或"summarized”。
- 在complete操作中。将中心对数比 (clr) 转换直接应用于细菌物种水平和代谢物簇水平丰度。
- 在Summarized操作中,将细菌物种级丰度聚合到属级丰度中,将代谢物聚类级丰度聚合到类级丰度中(从综合分类信息系统(ITIS)检索到物种到属的转换表,以及从 Franzosa 等人的补充中检索到代谢物到物种的转换表)。
4、神经编解码(NED)网络模型处理
4.1 神经编解码(NED)网络
神经编码器解码器 (NED) 框架通过潜在特征空间,利用来自另一个多变量过程的信息预测多变量随机过程。从输入(即微生物)中提取相关信息到潜在空间的网络部分称为编码器。预测潜在空间输出(即代谢物)的网络部分称为解码器。潜在特征空间是编码器和解码器之间的隐藏层。与直线回归等直接线性模型相比,编码器解码器网络的权重会经过非线性激活,因此具有更强健的表示形式,理论上NED的结果会表现得更好。
本文为了保持模型的可解释性,并使其对少量训练数据更加可靠,将隐藏层的数量限制为一层。文章初始实验表明,大量隐藏层并不能提高 NED 的预测性能,因为很容易在有限的训练数据上过度拟合。
在运行中,模型的两个部分按顺序协同工作:
(1)编码器从微生物X中提取相关信息,通过一个编码函数存储在潜在变量Z中。
(2)解码器使用解码函数解密Z中的潜在内容以预测Y(代谢组丰度)的值。
4.2 网络的稀疏化
为了进一步提高模型的稳定性和可解释性,文章试图学习一个具有最少活动权重的NED模型:一个大多数权重等于零的稀疏神经网络。本文使用了一种训练前筛选方法:(1)筛选阶段,找出从微生物丰度预测代谢物丰度提取所需信息最有用的连接。其他所有的连接标记为不必要的链接。(2)训练阶段,网络在前向和后向操作中停用这些多余的链接。
4.3 非负权重约束
与完全连接的 NED 相比,稀疏编解码网络具有少量的活动连接,许多连接自发地形成负权重值。神经网络中的这些负连接会对分析层与层之间的影响关系产生阻碍。因此,本文为了提高网络的可解释性,使用非负约束来防止权重降至零以下。方法是在每次训练迭代时将参数的中间估计值限制在[ 0,∞]。
5、模型评估
为了深入理解细菌-代谢物的关系,文本想要训练一个不仅准确,而且高度可解释的模型,其性能在不同方面是稳定的。因此本文应用用于评估预测模型的三个标准:准确性、稀疏性和稳定性。
5.1 准确性
预测模型的准确性是由预测的代谢物丰度与测量的代谢物丰度的皮尔逊相关系数来计算的。为了减轻虚假相关性的影响,相关性总是使用 clr 转换的数据计算。对于所有实验,本文使用五倍交叉验证方案,在五个不同的80%-20% 训练测试集上拆分来验证模型,并报告平均准确性。
5.2 稀疏性
模型中的活动线性连接的数量可以衡量模型的稀疏性。
5.3 稳定性
虽然准确性和稀疏性是评价预测模型性能的理想属性,但它们的性能只有在训练集构成的变化中保持一致的时候,才能说明是可靠的。本文通过测量不同训练集中模型参数的平均对等相似性来评估预测模型的稳定性,将数据集分为 5 份,学习每份的模型实例。然后,对于每对模型实例,计算模型参数之间的 Pearson相关系数。
本文使用模型中每个层之间的连接的二元邻接矩阵来计算稳定性指数来模型架构的稳定性。对于完全连接的模型(例如,线性回归或 CanCor) 任意一层中的每一个因子都会影响后续层中的所有因子,稳定性指数总是等于一,所以是没有意义的。然而,对于稀疏神经编码网络而言,稳定性指数可以衡量跨层连接的一致性和可靠性。对于 NED 及其变种等多层模型,作者使用第一个模型实例初始化其他实例的训练,以便保留中层变量的对应性。
6、结果和讨论
作者对比了一些线性模型,对比作者提出的神经编解码网络与其他模型性能在准确性、稀疏性和稳定性方面的优劣。
从微生物组(物种水平)丰度预测代谢物(集群水平)丰度时的性能如下图。
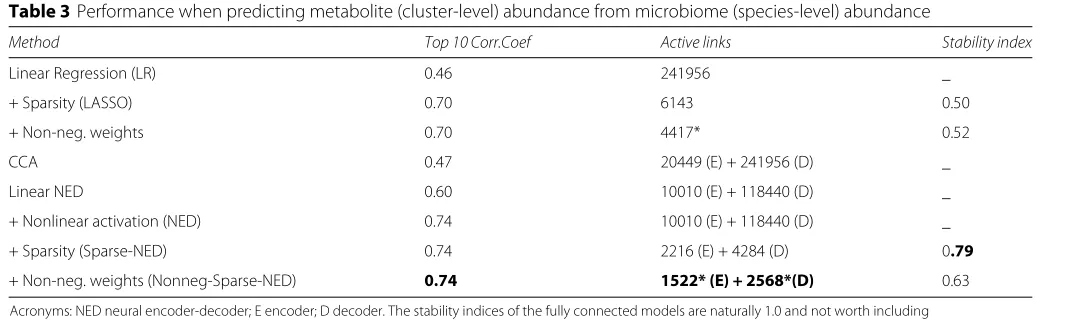
图2. complete数据集评估
预测微生物群(属级)丰度的代谢物(功能类别)丰度时的性能如下图。
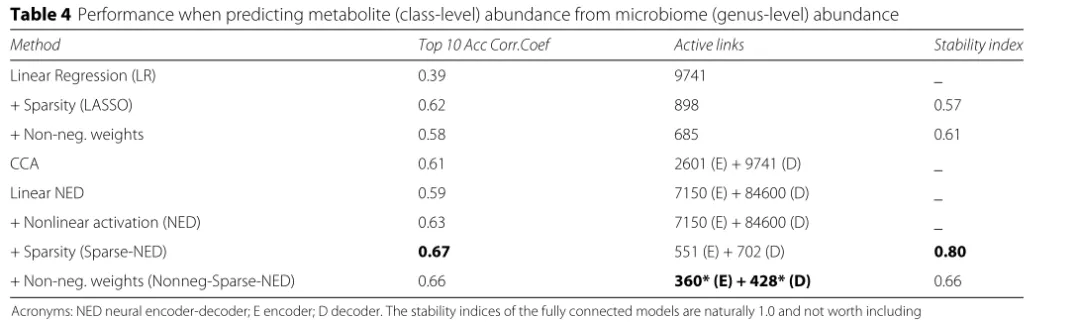
图3. summarized数据集评估
炎症性肠病(IBD)给发达国家带来了沉重的健康负担。虽然IBD没有传染性,但克罗恩病(CD)和溃疡性结肠炎(UC)患者会表现出肠道微生物群异常以及肠道代谢情况改变的症状。这篇文章提出了一个神经编码器解码器模型,以学习一组加权连接,这些连接可以通过微生物的丰度来预测代谢物的丰度。并且,本文表明,作者提出的神经编码器解码器网络是优于用于微生物群代谢预测的线性模型,并且对网络的稀疏化以及非负权重约束等操作,进一步提高了编码器解码器模型的准确性、稳定性和可解释性。更重要的是,神经编码器解码器模型不仅仅是一个最大限度地提高预测精度黑匣子,而且模型的隐藏层可以帮助可视化微生物和代谢物之间的预测关系。
参考资料
- 2020, Deep in the Bowel: Highly Interpretable Neural Encoder-Decoder Networks Predict Gut Metabolites from Gut Microbiome。 https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-6652-7
- https://mp.weixin.qq.com/s/BVOPQQQ_56MZ-knsWHKdDw
