【2.3.1】CNVkit
胚细胞拷贝数变异(CNV)和体细胞拷贝数变异(SCNA)在各种系统性综合征和癌症中具有重要意义。尤其在癌症研究领域,CNV已被多项研究证明参与了癌症的发生和发展,其数量和复杂度是很多疾病非常重要的诊断指标。随着高通量测序技术的成熟,可以根据测序reads的序列深度变化来推断基因组的拷贝数信息。CNVkit就是专门开发用于全基因组尺度拷贝数检测的一款软件。该软件的一大特点就是同时使用捕获的靶序列reads和非特异性的脱靶序列reads来推断分布在整个基因组中的基因拷贝数。
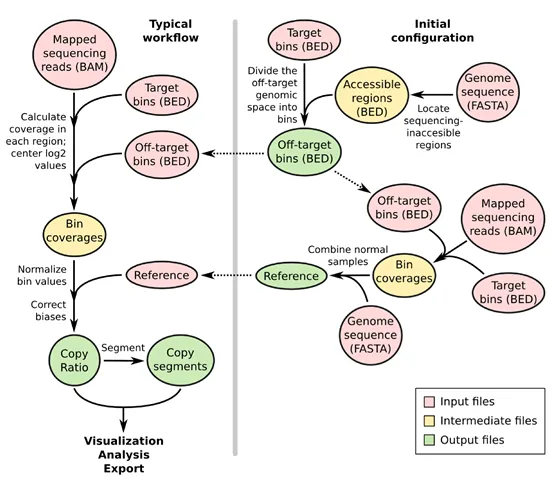
CNVkit使用捕获的靶标reads和非特异性捕获的靶标外的reads来计算每个样品在基因组中的拷贝比率(log2)。简单说就是,根据靶标区域之间的基因组位置来调配脱靶集合。然后,将靶标区和靶标外的位置分别用于计算每个间隔内的平均reads深度。再将靶标区和靶标外的reads深度进行组合,并根据对照样本得出的参考值进行归一化,并同时针对多个系统偏差进行校正,以得出最终的拷贝比率(log2)表。接着基于比率值(log2)运行内置的分割算法,推断离散拷贝数分段情况。最后对结果进行可视化并作进一步分析。
一、cnvkit 安装
官网:https://cnvkit.readthedocs.io/en/latest
github地址: https://github.com/etal/cnvkit/
#方法1
wget -c https://github.com/etal/cnvkit/archive/v0.9.7.zip
unzip v0.9.7.zip
cd ~/cnvkit-0.9.7
python3 ~/cnvkit-0.9.7/cnvkit.py -h
#方法2
pip3 install cnvkit
#指定安装目录
pip3 install "cnvkit==0.9.7" --prefix="~/CNVkit-v0.9.7"
#方法3:获得python 安装包,再用pip安装
wget https://files.pythonhosted.org/packages/3e/86/13f74ee3d3daccc6dc29420a5ed55dec1263991998ebd43e20c3e3b2a397/CNVkit-0.9.8.tar.gz
#依赖的相关模块
biopython>=1.62
pomegranate>=0.9.0
matplotlib>=1.3.1
numpy>=1.9
pandas>=0.23.3
pyfaidx>=0.4.7
reportlab>=3.0
scikit-learn
scipy>=0.15.0
networkx>=2.0
pysam
二、cnvkit 使用
2.1 CNV拷贝数检测(Copy number calling pipeline)
注意事项:
- 除了WGS检测外,都需要准备BED格式文件;
- 最好使用 Tumor-Normal 配对分析,如果没有Tumor,建议使用多个正常人germline样本作为对照;
0.使用命令batch 进行 CNV 分析
a) Tumor-Normal 配对样本
# From baits and tumor/normal BAMs
python3 cnvkit.py batch \
sample.Tumor.bam \
--normal sample.Normal.bam \
--targets panel.bed \
--fasta hg19.fasta \
--annotate $cnvkit_path/data/refFlat.txt \
--access $cnvkit_path/data/access-5kb-mappable.hg19.bed \
--output-reference my_reference.cnn \
--output-dir results \
--diagram --scatter
b) Tumor Only 样本,无配对样本,此使需要用其他基线 Normal 样本创建 reference.cnn文件,没有Normal样本也要无条件创造reference.cnn 文件,因为 –normal 和 –reference 两个参数必须选择一个
第一步:生成基线 reference.cnn 文件
# Reusing targets and antitargets to build a new reference, but no analysis
python3 cnvkit.py batch \
--normal *Normal.bam \
--fasta hg19.fasta \
--access $cnvkit_path/data/access-5k-mappable.hg19.bed \
--targets panel.target.bed \
--antitargets panel.antitarget.bed \
--output-reference normal.reference.cnn
参数说明:
--normal:基线样本BAM文件,空分割
--fasta:参考基因组文件
--access:可比对区域文件(bin:5kb)
--targets :panel 内 区域文件,可有 target 命令获得
--antitargets:off-panel 区域文件,可有 antitarget 命令获得
第二步:肿瘤样本 call cnv
# Reusing a reference for additional samples
python3 cnvkit.py batch \
sample.bam \
--reference normal.reference.cnn \
--output-dir results/
参数说明:
--reference :上一步生成的 baseline 的 depth、log2 等文件
--output-dir : 输出目录,默认是当前目录
2.2 以下是batch的拆解步骤
1.准备BED格式文件,格式如下,必须要有第四列基因名称
chr1 1508981 1509154 SSU72
chr1 2407978 2408183 PLCH2
chr1 2409866 2410095 PLCH2
a)若你的BED 区域太大,需要进行分割,可用 target 运行
python3 cnvkit.py target \
panel.bed \
--split \
--output panel.target.bed
–split :默认是267bp,因为人的exon大小平均是200bp。bins数据越少,噪音越大,检测的分辨率越低。因此建议使用该参数对BED区域进行重新调整。也可以通过设置 –avg-size 进行设置 bin 的大小,默认是 200 / 0.75 ≈ 267,但实际上生成的BED region 最大值 <= avg_size150%,最小值 >= avg_size * 75%
b)若你的BED文件没有基因名称,可使用 target –annotate 参数进行注释,需要去UCSC上下载注释文件:
# 下载 refFlat.txt
wget http://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/refFlat.txt.gz
#$cnvkit_path 是cnvkit安装目录
python3 cnvkit.py target \
panel.bed \
--annotate $cnvkit_path/data/refFlat.txt \
--split \
--output panel.target.bed
c)若你没有BED文件,可以尝试在这里下载 Astra-Zeneca’s reference data repository 或者可以用 script guess_baits.py 进行创建(还未好好研究用法
2. 使用命令 access< 确定测序手段无法测到的区域(这些主要是着丝粒,端粒和高度重复的区域,在参考基因组序列中,这些区域充满了大段“ N”字符) (一般不需要做,有做好的文件)
python3 cnvkit.py access \
hg19.fa \
--output access-excludes.hg19.bed
3. 使用命令 antitarget 获得 off-targt BED 区域
python3 cnvkit.py antitarget \
panel_out.split.bed \
--access data/access-5k-mappable.hg19.bed \
--output panel.antitarget.bed
–access data/access-5kb-mappable.hg19.bed : hg19基因组上可进行测序的区域,随软件下载存放在data目录下的文件
4. 使用命令 autobin 计算BAM文件 target-BED 和 off-target-BED 平均测序深度和bins数量
python3 cnvkit.py autobin \
*.bam \
--method hybrid \
--access data/access-5k-mappable.hg19.bed \
--targets panel.bed
### 输出结果
Depth Bin size
Target: 3059.880 33
Antitarget: 0.099 500000
### 还会输出2个BED文件 panel.target.bed panel.antitarget.bed
参数:
--method {hybrid,amplicon,wgs} : 测序方法,包括杂交捕获、扩增子、WGS测序
--targets : 待评估的BED文件
--access 可进行测序的基因组序列
5. 使用命令 coverage 计算BAM文件覆盖度
1)对该样本,按照BED区域,统计target 和 off-target 每个区域的depth 和 log2 ,输入BAM文件要求:
- BAM file must be sorted by coordinates
- If BAM has not .bai, it will Indexing it
- If BAM is old than BAM.bai, it will Indexing it
- Use pysam module creat BAM.bai
2)在计算Depth时会对BAM进行如下过滤:
-
read.is_duplicate will be filtered
-
read.is_secondary will be filtered
-
read.is_unmapped will be filtered
-
read.is_qcfail will be filtered
-
read.mapq < min_mapq will be filtered 默认 min_mapq = 0,且无法传参
#第一步 target coverage
python3 cnvkit.py coverage
sample.bam
panel.targets.bed
–output sample.targetcoverage.cnn
#第二步 off-target coverage
python3 cnvkit.py coverage
sample.bam
panel.antitargets.bed
–output sample.antitargetcoverage.cnn
生成的文件格式如下
chromosome start end gene depth log2
chr1 11166661 11166959 MTOR 3651.15 11.8341
chr1 11166959 11167258 MTOR 3633.48 11.8271
chr1 11167258 11167557 MTOR 3134.66 11.6141
chr1 11168237 11168343 MTOR 2292.16 11.1625
chr1 11169346 11169427 MTOR 2238.02 11.128
6. 使用命令 reference合并 targetcoverage.cnn 和 antitargetcoverage.cnn,生成reference.cnn 文件 并计算区域GC含量和 repeat 区域比例
a)一个样本,实际上1个样本不需要这步操作了:
python3 cnvkit.py reference \
sample.targetcoverage.cnn sample.antitargetcoverage.cnn \
--fasta ucsc.hg19.fa \
--output sample.reference.cnn
b) 多个样本,生成 基线 reference.cnn
python3 cnvkit.py reference \
*coverage.cnn \
--fasta ucsc.hg19.fa \
--output normal.reference.cnn
coverage.cnn 后缀必须是 targetcoverage.cnn 和 antitargetcoverage.cnn,会根据后缀进行区分 target和 off-target
生成文件格式如下:
chromosome start end gene log2 depth gc rmask spread
chr1 10500 176917 Antitarget 0.345899 0.0253011 0.42974 0.479687 0.44347
chr1 227917 267219 Antitarget 0.666802 0.0379031 0.392423 0.515623 0.728676
chr1 318219 470868 Antitarget 0.0667445 0.0172231 0.425827 0.561189 0.400429
chr1 521868 563949 Antitarget -0.409377 0.0155651 0.495568 0.471377 0.825082
若没有对照样本,可使用 reference 生成 og2=0.0的 reference.cnn 文件:a “dummy” reference
python3 cnvkit.py reference \
--fasta ucsc.hg19.fa \
--targets targets.bed \
--antitargets antitargets.bed \
--output FlatReference.cnn
生成文件:
chromosome start end gene log2 depth gc rmask spread
chr1 10500 176917 Antitarget 0 1 0.42974 0.479687 0
chr1 227917 267219 Antitarget 0 1 0.392423 0.515623 0
chr1 318219 470868 Antitarget 0 1 0.425827 0.561189 0
7. 使用命令 fix 对 depth 进行 偏差纠正,需要同时输入 targetcoverage.cnn、antitargetcoverage.cnn 和 reference.cnn,生成 cnr(copy number segments)文件
/data/CAP/Software/Python3/Python-v3.7.0/bin/python3 \
/data/CAP/Software/CNVkit/cnvkit-0.9.7/cnvkit.py \
fix \
sample.targetcoverage.cnn \
sample.antitargetcoverage.cnn \
sample.reference.cnn \
--output sample.cnr
参数说明:
sample.targetcoverage.cnn: tumor 样本 target cnn结果 sample.antitargetcoverage.cnn : tumor 样本 off-target cnn 结果 sample.reference.cnn:基线样本reference.cnn 结果
输出文件:
chromosome start end gene log2 depth weight
chr1 10500 176917 Antitarget -0.0131375 0.0135142 0.99188
chr1 227917 267219 Antitarget 0.571298 0.030431 0.309595
8.使用命令 segment从cnr文件中推断出离散的拷贝数段
python3 cnvkit.py segment sample.cnr --output sample.cns
9.使用命令call 计算出每个分段的绝对整数拷贝数
python3 cnvkit.py call sample.cns -o Sample.call.cns
python3 cnvkit.py call sample.cns -y -m threshold -t=-1.1,-0.4,0.3,0.7 -o Sample.call.cns
python3 cnvkit.py call sample.cns -y -m clonal --purity 0.65 -o Sample.call.cns
python3 cnvkit.py call sample.cns -y -v Sample.vcf -m clonal --purity 0.7 -o Sample.call.cns
10. cnr cns 结果解读
1.sample.cnr title
chromosome start end gene depth log2 weight
2.sample.cns title
chromosome start end gene log2 depth probes weight ci_lo ci_hi
3)sample.call.cns title
chromosome start end gene log2 cn depth p_ttest probes weight
2.2 CNV拷贝数可视化(Plots and graphics)
1.使用 scatter 根据Log2值进行结果可视化
python3 cnvkit.py scatter sample.cnr -s sample.cns
2.使用diagram根据gain或者loss进行结果可视化
cnvkit.py diagram sample.cnr
cnvkit.py diagram -s sample.cns
cnvkit.py diagram -s sample.cns sample.cnr
3.使用heatmap 进行多个样本间的结果可视化
python3 cnvkit.py heatmap *.cns
2.3(Text and tabular reports)
1.使用breaks
2.使用genemetrics
python3 cnvkit.py genemetrics sample.cnr
python3 cnvkit.py genemetrics sample.cnr -s sample.cns -t 0.4 -m 5 -y
3.使用sex
4.使用metrics
5.使用segmetrics
参考资料
