【2.5.1】GATK-HaplotypeCaller
GATK HaplotypeCaller由于其准确性高已成为胚系短变异检测的行业标准,
一、GATK-HaplotypeCaller简介
基因组变异检测是基因组学领域一个非常重要的问题,是遗传性疾病溯源,物种进化等分析的前提。而目前最主流、使用最广泛的变异检测软件当属 Broad Institute 开发的 GATK(Genome Analysis ToolKit) 组件。GATK 设计之初是用于分析人类的全外显子和全基因组数据,随着不断发展,现在也可以应用于其他的物种。
GATK官网提供了一整套完整的变异检测分析流程:GATK Best Practices。如下
图示:
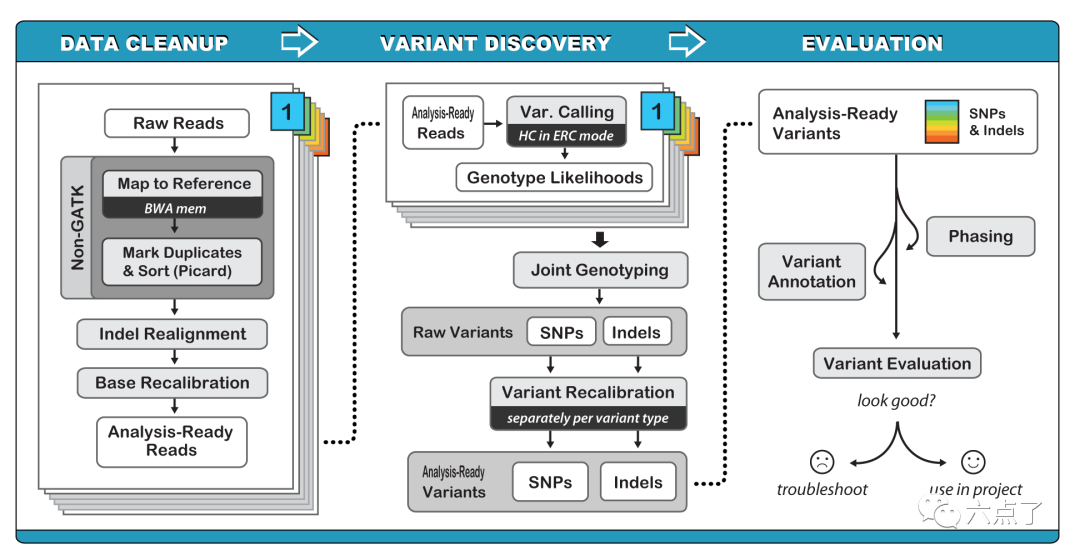
其中,HaplotypeCaller 是 GATK 检测变异(SNP/INDEL)的核心模块,主要通过单倍型的局部重组来实现准确的 SNP 和 INDEL 检测。 GATK-HaplotypeCaller参考网址[1]
HaplotypeCaller 能够从头组装变异活跃的区域(active region)并进行 SNP/INDEL检测。即每当程序遇到显示出变异迹象的区域时,它就会丢弃现有的比对信息并完全重新组装该区域中的reads。因此 HaplotypeCaller 在一些传统方法难以检测的区域上会更加准确,也使得在检测 indel 方面比 UnifiedGenotyper(gatk旧版变异检测模块) 等基于位置的变异检测工具效果更好。
HaplotypeCaller 能够处理非二倍体生物以及混合的实验数据,还能够正确处理可变剪切,可以适用 RNAseq数据。
二、 GATK-HaplotypeCaller的变异检测的基本原理
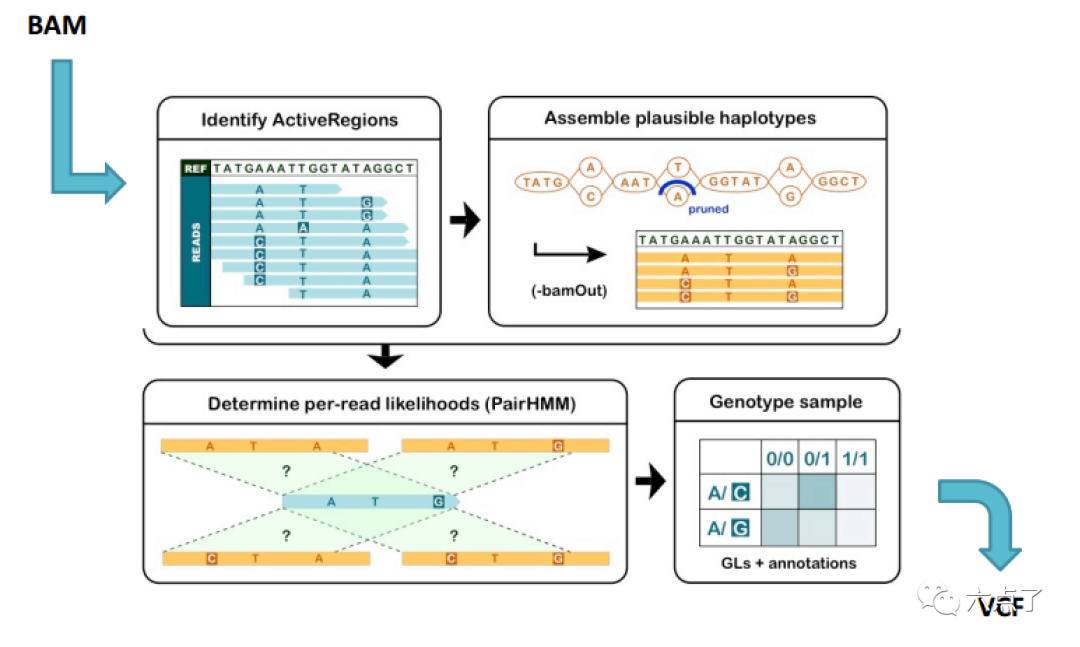
GATK-HaplotypeCaller 模块进行 SNP/indel 检测的基本工作流程包含四个主要步骤:
-
识别活跃区域
-
通过重组装活跃区域确定单体型
-
确定每个read的单倍型的似然值
-
确定基因型。
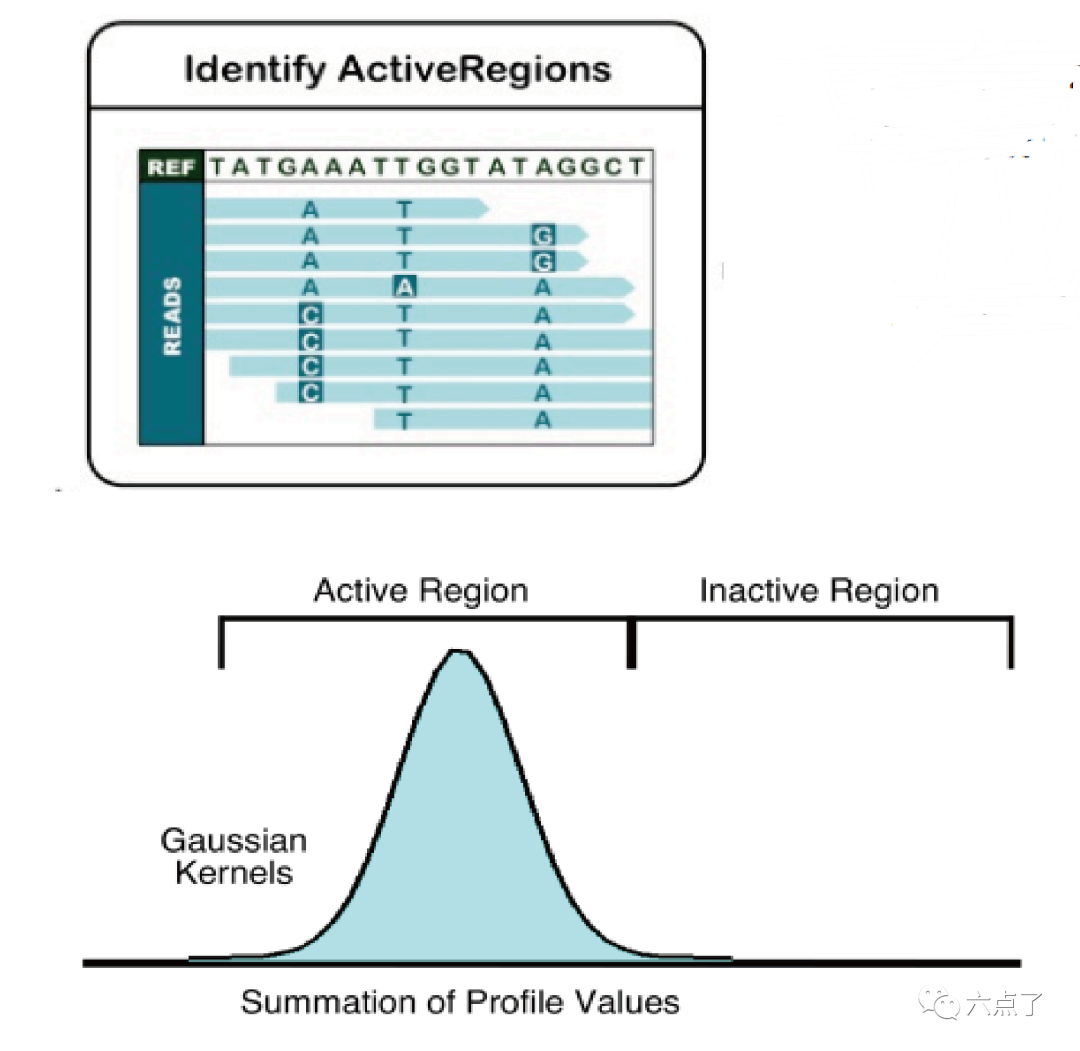
2.1 识别活跃区域
沿着参考基因组以一定的窗口滑动,统计比对的 mismatches, indels 和 softclips等信息计算基因组每个位置的活跃得分,使用平滑算法进行处理,此处相当于测定该区域熵值。当熵值达到某个设定的阈值时即确定该区域为active region,用于后续组装。
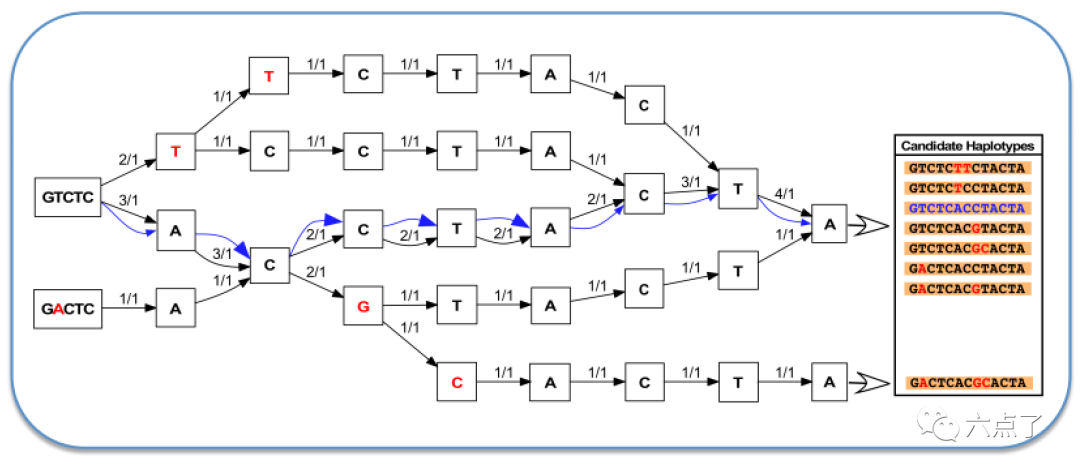
2.2 通过重组装活跃区域确定单体型
对于每个活动区域,忽略之前的read比对结果,重新利用该区域的reads构建一个类似 De Bruijn 的图来组装活跃区域并识别数据中可能存在的单倍型。然后,使用 Smith-Waterman 算法将每个单倍型与参考单倍型重新对齐,以识别潜在的变异位点。如下图示:最优的路径通过构图的方式进行打分,得到候选的单体型路径。
2.3 确定每个read的单倍型的似然值
对于每个活动区域,程序使用 PairHMM 算法将每个read与每个单倍型进行成对比对, 产生一个单倍型似然值矩阵。然后将这些似然值边缘化以获得给定reads的每个潜在变异位点的等位基因可能性。
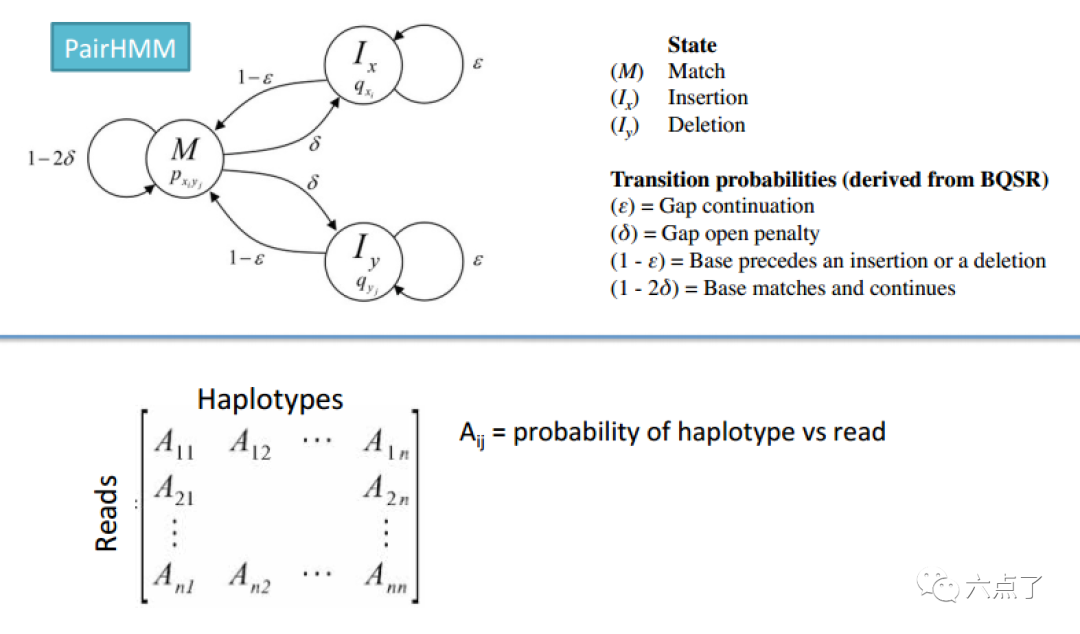
2.4 确定基因型
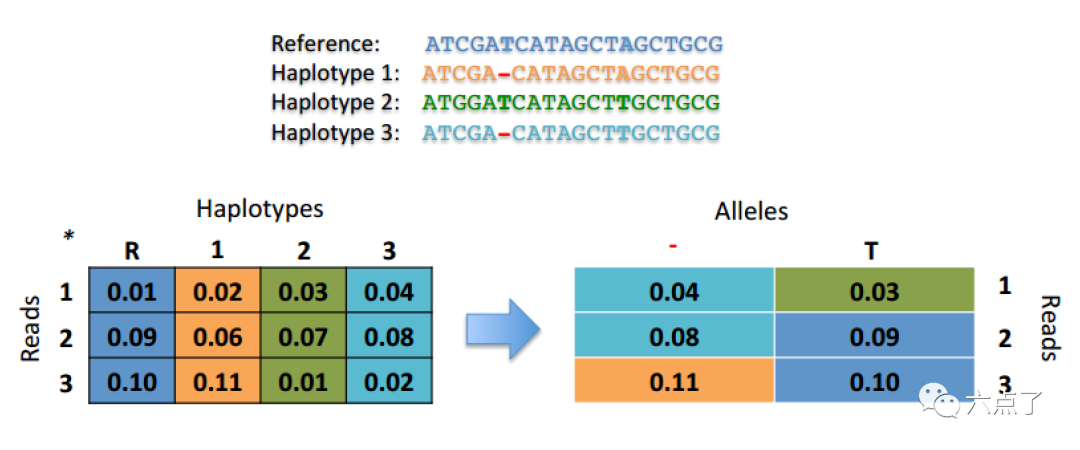
将前面 PairHMM 步骤得到的候选单倍型的似然值应用贝叶斯算法转化为每个位点基因型的似然值,如下图所示:
三、 GATK-HaplotypeCaller的安装和使用
目前GATK已更新到GATK4。和GATK3相比,GATK4在算法上进行了优化,运行速率有所提高,而且整合了picard 软件的功能。github地址[2].
3.1. 安装
GATK软件运行有两种方式一种是运行通过java 调用jar包运行,一种是使用gatk脚本文件,此时需要安装Python 2.6以上版本,不管是哪种方式,都需要机器上安装java 8或以上版本。
wget https://github.com/broadinstitute/gatk/releases/download/4.1.5.0/gatk-4.1.5.0.zip
unzip gatk-4.1.5.0.zip
注意此处下载最好是直接下载gatk-**.zip包,而不要下载Source code (zip) 和 Source code (tar.gz)两个源码包,不然还需要重新build gatk。当然如果你的环境无法支持那就另说了。下载完解压即可看到可执行程序gatk, 以及对应的jar包,可以运行./gatk –list测试下是否可运行。
3.2. 使用说明
运行./gatk HaplotypeCaller -h 查看 HaplotypeCaller 的参数细节,详细说明到官网查看会更清晰一些https://gatk.broadinstitute.org/hc/en-us/articles/360040096812-HaplotypeCaller
尽管HaplotypeCaller官网参数非常多,但实际用上的却不多,大部分按默认参数即最佳,这里列举常用参数进行说明:
--input -I [] # BAM/SAM/CRAM file containing reads 指定输入的bam、sam、cram文件
--output -O null # File to which variants should be written 指定输出vcf名字
--reference -R null # Reference sequence file 指定参考序列,需要和比对时候使用的参考基因组一致
--intervals -L [] #One or more genomic intervals over which to operate 指定变异检测的区间,可以是bed文件也可以是染色体名字
--emit-ref-confidence -ERC NONE # Mode for emitting reference confidence scores
包含以下三种变异检测模式:
#1. NONE:只记录变异位点
#2. BP_RESOLUTION:记录变异和非变异位点,每个位点信息都展示
#3. GVCF:记录变异和非变异位点,其中非变异位点以block块展示
--pcr-indel-model CONSERVATIVE #The PCR indel model to use设置针对PCR indel的矫正模型,包含以下几种模式
#1. NONE:不会应用专门的 PCR 错误模型;如果存在碱基插入/删除质量,它们将被使用
#2. HOSTILE:将应用一个最激进的模型,该模型会牺牲真阳性以消除更多的假阳性
#3. AGGRESSIVE:将应用相对激进的模型,牺牲真阳性以消除更多的假阳性
#4. CONSERVATIVE:将应用一个不那么激进的模型,该模型试图以允许更多误报为代价来保持较高的真阳性率
注意:
1)对于队列分析,通常会使用 -ERC GVCF 参数来生成gvcf文件以支持下游多样本联合分析 2)对于PCR-free的样本需要使用-pcr_indel_model NONE取消PCR indel模型。
运行命令如下:
gatk --java-options "-Xmx4g" HaplotypeCaller \
-R Homo_sapiens_assembly38.fasta \
-I input.bam \
-O output.g.vcf.gz \
-ERC GVCF \
-L chr1
四、GATK-HaplotypeCaller实战
下面我们找个NA12878样本的bam 文件数据,具体来实践一下吧。
4.1 数据下载
下载运行所需的数据:参考基因组及其索引,bam文件
wget http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/Reference/hg19.chrM.fasta_BwaMem_index/hg19.chrM.fasta
wget http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/Reference/hg19.chrM.fasta_BwaMem_index/hg19.chrM.dict
wget http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/Reference/hg19.chrM.fasta_BwaMem_index/hg19.chrM.fasta.fai
wget http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/WGS/hg19.chrM.bwa-mem.sort.rmdup.BQSR.bam
wget http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/WGS/hg19.chrM.bwa-mem.sort.rmdup.BQSR.bai
4.2 运行命令
此处我们使用Gvcf模式,且只检测chrM的变异
gatk --java-options "-Xmx4g" HaplotypeCaller \
-R hg19.chrM.fasta \
-I hg19.chrM.bwa-mem.sort.rmdup.BQSR.bam \
-O chrM.g.vcf.gz \
-ERC GVCF \
-L chrM
4.3 输出结果
运行完即可看到chrM.g.vcf.gz文件的生成。
此外,我们的sixoclock官网基于CWL (common workflow language) 对 GATK-HaplotypeCaller 软件进行了封装,通过我们开发的sixbox 软件可以快速进行软件的运行。对sixbox不了解可以通过六点了官网了https://docs.sixoclock.net/clients/sixbox-linux.html解下。下面是具体的运行步骤如下:
1)下载cwl 源码 sixbox pull 2e932c25-8c36-4733-bb91-79a137282013 或 点击下载按钮下载 gatk_HaplotypeCaller.cwl https://www.sixoclock.net/application/pipe/2e932c25-8c36-4733-bb91-79a137282013
-
使用sixbox生成参数模板文件(YAML) , 并配置yaml文件
sixbox run –make-template ./HaplotypeCaller.cwl > HaplotypeCaller.job.yaml vim HaplotypeCaller.job.yaml # 编辑参数配置文件,替换或设置参数以实现个性化分析
可以直接粘贴下方示例内容到HaplotypeCaller.job.yaml
reference: # type "File"
class: File
path: http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/Reference/hg19.chrM.fasta_BwaMem_index/hg19.chrM.fasta
secondaryFiles:
- class: File
path: http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/Reference/hg19.chrM.fasta_BwaMem_index/hg19.chrM.dict
- class: File
path: http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/Reference/hg19.chrM.fasta_BwaMem_index/hg19.chrM.fasta.fai
output_vcf: chrM.g.vcf.gz # type "string"
#java_options: '-Xmx6g -XX:ParallelGCThreads=4' # type "string"
intervals: # array of type "string" (optional)
- "chrM"
input_bam: # array of type "File"
class: File
path: http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/WGS/hg19.chrM.bwa-mem.sort.rmdup.BQSR.bam
secondaryFiles:
- class: File
path: http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/WGS/hg19.chrM.bwa-mem.sort.rmdup.BQSR.bai
ERC: 'GVCF' # type "string"
-
使用sixbox运行
sixbox run ./HaplotypeCaller.cwl ./HaplotypeCaller.job.yaml
#或者指定输出目录 sixbox run –outdir /home/path ./HaplotypeCaller.cwl ./HaplotypeCaller.job.yaml
运行结果即可看到当前目录或者指定的输出目录输出chrM.g.vcf.gz结果。
以上为我们给大家带来的宏基因组组装工具Megahit基本原理知识,以及运行详细操作过程。
如果对生物医疗健康大数据相关内容感兴趣也可以持续关注我们。想要探索更多生物医疗大数据分析工具和软件的介绍和使用请看六点了官网[3]。
五、我的案例
gatk HaplotypeCaller -R /opt/ref/ref.fasta -I ${output}/${ID}/${ID}.bam -O ${output}/${ID}/${ID}.vcf --minimum-mapping-quality 10 --ploidy 2 --annotation ChromosomeCounts --annotation FisherStrand --annotation StrandOddsRatio --annotation QualByDepth --annotation ReadPosRankSumTest
参考资料
