【2.3.7】使用 Manta 检测结构变异
Manta 是 illumina 公司开发的一款用于检测结构变异 Structural Variant 和 Indel 的软件,Manta 检测 SV 和 Indel 分为两个主要步骤:
- 扫描基因组以找到SV相关区域,
- 分析,评分和输出在这些区域发现的SV。其输出的结果可以作为 strelka 的输入,以提高 strelka 对 indel 检测的准确性。
值得注意的事,该软件既可以用于 germline SV 的检测(家系样本),也可以用于 Somatic SV 的检测(肿瘤样本)
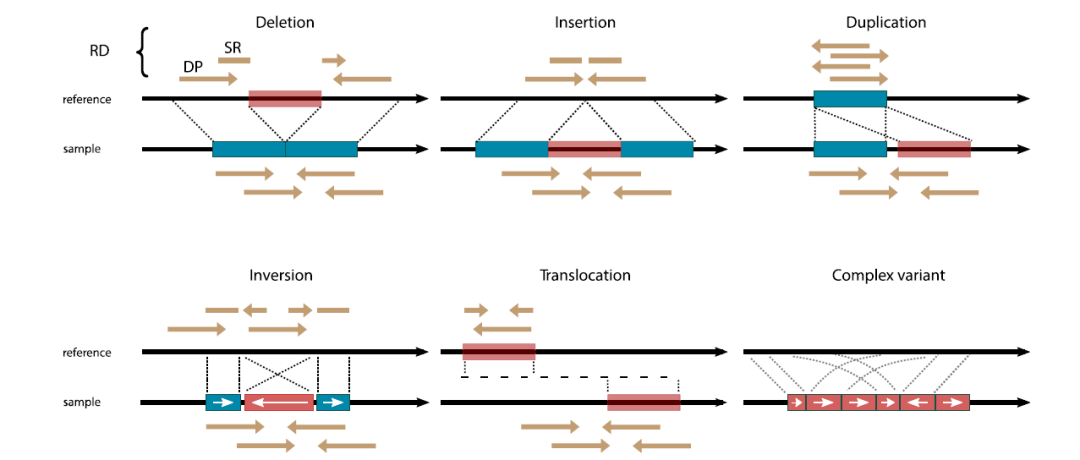
二、下载安装
该软件支持多种安装方式,如果要从源码安装,则需要解决环境问题如:python 2.6+、gcc 4.8+ 等,不推荐。开发团队提供了二进制版本,下载解压后即可使用,不过是面向 Centos 系统的(感兴趣的读者可以尝试在Ubuntu上使用这种方式安装)
wget -c https://github.com/Illumina/manta/releases/download/v1.6.0/manta-1.6.0.centos6_x86_64.tar.bz2
tar -jxvf manta-1.6.0.centos6_x86_64.tar.bz2
Ubuntu 系统的软件的安装方法是(注意,编译的时候要在python 2的环境下,另外需要修改–prefix 参数指定安装路径):
wget -c https://github.com/Illumina/manta/releases/download/v1.6.0/manta-1.6.0.release_src.tar.bz2
tar -xjf manta-1.6.0.release_src.tar.bz2
cd manta-1.6.0.release_src/
mkdir build && cd build
# Ensure that CC and CXX are updated to target compiler if needed, e.g.:
# export CC=/path/to/cc
# export CXX=/path/to/c++
../manta-1.6.0.release_src/configure --jobs=4 --prefix=${HOME}/biosoft/manta
make -j4 install
由于依赖的问题,安装比较麻烦,经常安装失败。或者可以考虑用conda安装。安装好了之后,可以使用下面命令检查一下(注意是python2):
python ${MANTA_INSTALL_PATH}/bin/runMantaWorkflowDemo.py
实际运行
运行过程分为两步:
- 配置 config
- 运行脚本:
对于 germline SV 的示例:
${MANTA_INSTALL_PATH}/bin/configManta.py \
--bam sample.bam \
--referenceFasta Homo_sapiens_assembly38.fasta \
--runDir ${MANTA_ANALYSIS_PATH}
${MANTA_ANALYSIS_PATH}/runWorkflow.py
如果是家系样本,可以同时输入多个bam文件:
${MANTA_INSTALL_PATH}/bin/configManta.py \
--bam sample1.bam \
--bam sample2.bam \
--bam sample3.bam \
--referenceFasta Homo_sapiens_assembly38.fasta \
--runDir ${MANTA_ANALYSIS_PATH}
${MANTA_ANALYSIS_PATH}/runWorkflow.py
对于 Somatic (也支持非配对的肿瘤样本,但是运行方法不一样):
${MANTA_INSTALL_PATH}/bin/configManta.py \
--normalBam normal.bam \
--tumorBam tumor.bam \
--referenceFasta Homo_sapiens_assembly38.fasta \
--runDir ${MANTA_ANALYSIS_PATH}
${MANTA_ANALYSIS_PATH}/runWorkflow.py
对于外显子测序,还可以使用 –callRegions指定 bed 文件,–exome 指定外显子测序。实际运行用到了下面的脚本:
#!/bin/sh
normal=$1
tumor=$2
mkdir 6.manta/${tumor}
${MANTA_INSTALL_PATH}/bin/configManta.py \
--tumorBam 5.gatk/${tumor}_bqsr.bam \
--normalBam 5.gatk/${normal}_bqsr.bam \
--referenceFasta ${GENOME} \
--callRegions ${bed} \
--exome \
--runDir 6.manta/${tumor} \
1>6.manta/${tumor}/${tumor}_manta.log 2>&1
python2 6.manta/${tumor}/runWorkflow.py -m local -j 4 1>6.manta/${tumor}/${tumor}_manta.log 2>&1
这样的话,每一个样本生成的文件会保存到单独的目录下,有:
$ ls 6.manta/*/
case1_biorep_A_techrep_1/:
case1_biorep_A_techrep_1_manta.log results/ runWorkflow.py* runWorkflow.py.config.pickle workflow.error.log.txt workflow.exitcode.txt workflow.warning.log.txt workspace/
case1_biorep_B/:
case1_biorep_B_manta.log results/ runWorkflow.py* runWorkflow.py.config.pickle workflow.error.log.txt workflow.exitcode.txt workflow.warning.log.txt workspace/
case1_biorep_C/:
case1_biorep_C_manta.log results/ runWorkflow.py* runWorkflow.py.config.pickle workflow.error.log.txt workflow.exitcode.txt workflow.warning.log.txt workspace/
case1_techrep_2/:
case1_techrep_2_manta.log results/ runWorkflow.py* runWorkflow.py.config.pickle workflow.error.log.txt workflow.exitcode.txt workflow.warning.log.txt workspace/
case2_biorep_A/:
case2_biorep_A_manta.log results/ runWorkflow.py* runWorkflow.py.config.pickle workflow.error.log.txt workflow.exitcode.txt workflow.warning.log.txt workspace/
case2_biorep_B_techrep_1/:
case2_biorep_B_techrep_1_manta.log results/ runWorkflow.py* runWorkflow.py.config.pickle workflow.error.log.txt workflow.exitcode.txt workflow.warning.log.txt workspace/
case2_biorep_C/:
case2_biorep_C_manta.log results/ runWorkflow.py* runWorkflow.py.config.pickle workflow.error.log.txt workflow.exitcode.txt workflow.warning.log.txt workspace/
case2_techrep_2/:
case2_techrep_2_manta.log results/ runWorkflow.py* runWorkflow.py.config.pickle workflow.error.log.txt workflow.exitcode.txt workflow.warning.log.txt workspace/
主要的结果是在 results 中,如:
$ ls case1_biorep_A_techrep_1/results/*
case1_biorep_A_techrep_1/results/evidence:
case1_biorep_A_techrep_1/results/stats:
alignmentStatsSummary.txt svCandidateGenerationStats.tsv svCandidateGenerationStats.xml svLocusGraphStats.tsv
case1_biorep_A_techrep_1/results/variants:
candidateSmallIndels.vcf.gz candidateSmallIndels.vcf.gz.tbi candidateSV.vcf.gz candidateSV.vcf.gz.tbi diploidSV.vcf.gz diploidSV.vcf.gz.tbi somaticSV.vcf.gz somaticSV.vcf.gz.tbi
输出结果
其中,主要是几个 vcf 文件:
-
diploidSV.vcf.gz : SVs and indels scored and genotyped under a diploid model for the set of samples in a joint diploid sample analysis or for the normal sample in a tumor/normal subtraction analysis. In the case of a tumor/normal subtraction, the scores in this file do not reflect any information from the tumor sample.
-
somaticSV.vcf.gz : SVs and indels scored under a somatic variant model. This file will only be produced if a tumor sample alignment file is supplied during configuration
-
candidateSV.vcf.gz : Unscored SV and indel candidates. Only a minimal amount of supporting evidence is required for an SV to be entered as a candidate in this file. An SV or indel must be a candidate to be considered for scoring, therefore an SV cannot appear in the other VCF outputs if it is not present in this file. Note that by default this file includes indels of size 8 and larger. The smallest indels in this set are intended to be passed on to a small variant caller without scoring by manta itself (by default manta scoring starts at size 50).
-
candidateSmallIndels.vcf.gz : Subset of the candidateSV.vcf.gz file containing only simple insertion and deletion variants less than the minimum scored variant size (50 by default). Passing this file to a small variant caller will provide continuous coverage over all indel sizes when the small variant caller and manta outputs are evaluated together. Alternate small indel candidate sets can be parsed out of the candidateSV.vcf.gz file if this candidate set is not appropriate.
对于 somatic SV ,得到的结果会保存在 somaticSV.vcf.gz 中:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT case1_germline case1_biorep_A_techrep_1
chr1 196910455 MantaDEL:2669:0:2:1:0:0 G <DEL> . PASS END=196914442;SVTYPE=DEL;SVLEN=-3987;CIPOS=0,4;CIEND=0,4;HOMLEN=4;HOMSEQ=GTTG;SOMATIC;SOMATICSCORE=85 PR:SR 26,0:61,0 72,10:128,40
不过,对于外显子数据,检测 SV 并不准确。于是我又用 WGS 的数据,跑了一遍 germline SV 的检测,diploidSV.vcf.gz 是:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT A_V300028149
chr1 778705 MantaINS:11:0:0:0:0:0 CA CGCCCTTGTGACGTCACGGAAGGCGCGCGCTTGCGACGTCACGGAAGGCGCGCCCTTGTGACGTCACGGAAGGCGCT 430 PASS END=778706;SVTYPE=INS;SVLEN=76;CIGAR=1M76I1D GT:FT:GQ:PL:PR:SR 0/1:PASS:249:480,0,246:0,0:17,13
chr1 789487 MantaDUP:TANDEM:10:601:606:0:0:0 G <DUP:TANDEM> 18 MinQUAL END=224014523;SVTYPE=DUP;SVLEN=223225036;IMPRECISE;CIPOS=-98,98;CIEND=-147,148 GT:FT:GQ:PL:PR 0/1:PASS:18:68,0,461:31,9
chr1 1068747 MantaDUP:TANDEM:36:1:2:0:0:0 C <DUP:TANDEM> 651 PASS END=1068825;SVTYPE=DUP;SVLEN=78;CIPOS=0,9;CIEND=0,9;HOMLEN=9;HOMSEQ=CCACGCGGG GT:FT:GQ:PL:PR:SR 0/1:PASS:84:701,0,81:4,0:23,27
chr1 1068824 MantaINS:36:2:2:0:0:0 G GGCCACGCGGGCTGTGCAGATGCAGGTGCGGCGGGGCGGGGCCACGCGGGCTGTGAAGGTGCAGGTGCGGCGGGGCAGA 999 PASS END=1068824;SVTYPE=INS;SVLEN=78;CIGAR=1M78I;CIPOS=0,10;HOMLEN=10;HOMSEQ=GCCACGCGGG GT:FT:GQ:PL:PR:SR 1/1:PASS:108:999,111,0:0,0:0,44
chr1 1207339 MantaDEL:49:0:1:0:0:0 GGAGACTGTCCTATGTCTTTCTGAGCCTCAGTTTCCCCTGTGGGCACCGAGGGGTTCTGGGACCCTGCCTCCACCAGGAAGCCTCCCTGGATTGCCCAGCCCTGCTTCTGCGCCGTCCAGCACAGGTGGAGACCCCCATGAATGCTGGGGGTGGGGGCTCTCGGGAACGTGAGCGTGGATGTGGTTCAACACCCTTTTGAGACCTGCAGCCACCGCCTCACCCCGTAAGGCGGTTCCTCCTTTTCCAAGGTAAATGACAGGAATTAGCTGTTTGTGACACCCCGGAGTTCTCAAATCCAAGATGTAGGAGCCTGCCTTGGAGAGGCAGCCCTCAGACACTGCAGAGAAGGAAGGGGTCTCTGCAGCTCCAGGCCGCCCCGACGCTCGGAAGGAAAGGGGTGGGGCCAGCTGGGCCTGGGGGC G 352 PASS END=1207760;SVTYPE=DEL;SVLEN=-421;CIGAR=1M421GT:FT:GQ:PL:PR:SR 0/1:PASS:352:402,0,643:21,6:41,12
chr1 15503064 MantaBND:929:0:1:0:0:0:1 C ]chr7:148936234]C 48 PASS SVTYPE=BND;MATEID=MantaBND:929:0:1:0:0:0:0;IMPRECISE;CIPOS=-64,65;BND_DEPTH=45;MATE_BND_DEPTH=42 GT:FT:GQ:PL:PR 0/1:PASS:48:98,0,228:16,8
chr7 148936234 MantaBND:929:0:1:0:0:0:0 T T[chr1:15503064[ 48 PASS SVTYPE=BND;MATEID=MantaBND:929:0:1:0:0:0:1;IMPRECISE;CIPOS=-132,133;BND_DEPTH=42;MATE_BND_DEPTH=45 GT:FT:GQ:PL:PR 0/1:PASS:48:98,0,228:16,8
......
Manta 得到的结构变异的 vcf 文件,和普通的 SNVs/INDELs 突变格式大致相同,但结构变异的 vcf 文件的第3列记录发生结构变异的类型:INS、DEL、DUP、BND
其中不同类型的变异记录方式略有不同,可以通过 vcf 文件第 3 列来判断发生变异的类型:
-
INDEL:如果检测到变异为 INDEL,片段长度一般不超过 1000 bp,且在 INFO 列还会记录 CIGAR值用以描述插入或者缺失的情况
-
BND:如果是 BND 事件,通常会有两个断点,为对应关系,记录在 INFO 列的 MATEID tag标签,即两条记录具有相同的 MATEID
-
Inversions:如果发生的变异是倒置,则会有 4 条记录,它们的 INFO 列中有共同的 EVENT标记,如以下示例:
chr1 205209478 MantaBND:9264:0:0:0:0:0:0 C CACAAC]chr1:205209646] 999 PASS SVTYPE=BND;MATEID=MantaBND:9264:0:0:0:0:0:1;SVINSLEN=5;SVINSSEQ=ACAAC;EVENT=MantaBND:9264:0:0:0:0:0:0;JUNCTION_QUAL=999;BND_DEPTH=46;MATE_BND_DEPTH=36 GT:FT:GQ:PL:PR:SR 1/1:PASS:174:999,177,0:0,12:0,32 chr1 205209503 MantaBND:9264:0:0:1:0:0:0 C [chr1:205209707[C 999 PASS SVTYPE=BND;MATEID=MantaBND:9264:0:0:1:0:0:1;CIPOS=0,41;HOMLEN=41;HOMSEQ=CAGAAATGGTGATAAAAATAAGATACTAAGATCATGACAGG;EVENT=MantaBND:9264:0:0:0:0:0:0;JUNCTION_QUAL=999;BND_DEPTH=42;MATE_BND_DEPTH=31 GT:FT:GQ:PL:PR:SR 1/1:PASS:174:999,177,0:0,11:0,20 chr1 205209646 MantaBND:9264:0:0:0:0:0:1 A AGTTGT]chr1:205209478] 999 PASS SVTYPE=BND;MATEID=MantaBND:9264:0:0:0:0:0:0;SVINSLEN=5;SVINSSEQ=GTTGT;EVENT=MantaBND:9264:0:0:0:0:0:0;JUNCTION_QUAL=999;BND_DEPTH=36;MATE_BND_DEPTH=46 GT:FT:GQ:PL:PR:SR 1/1:PASS:174:999,177,0:0,12:0,32 chr1 205209666 MantaBND:9264:0:0:1:0:0:1 C [chr1:205209544[C 999 PASS SVTYPE=BND;MATEID=MantaBND:9264:0:0:1:0:0:0;CIPOS=0,41;HOMLEN=41;HOMSEQ=TGTCATGATCTTAGTATCTTATTTTTATCACCATTTCTGGA;EVENT=MantaBND:9264:0:0:0:0:0:0;JUNCTION_QUAL=999;BND_DEPTH=31;MATE_BND_DEPTH=42 GT:FT:GQ:PL:PR:SR 1/1:PASS:174:999,177,0:0,11:0,20
另外,vcf 文件中的 INFO 列有很多 tag,不同的结构变异事件包含的列不同,每一个 tag 的信息是:
参考资料
