【2.3.6】Sequenza从WES推断出拷贝数概况
-
肿瘤 DNA 的外显子组或全基因组深度测序连同配对的正常 DNA 可能提供肿瘤特征的体细胞突变的详细图像。 然而,由于肿瘤标本中正常细胞的存在、瘤内或瘤体的异质性以及原始数据的庞大规模,这些序列数据的分析可能会变得复杂。
-
Sequenza是一个使用配对的肿瘤正常DNA测序数据来估计肿瘤细胞和倍性,并计算等位基因特异性拷贝数图谱和突变图谱的软件包。将Sequenza和两个以前发布的算法应用于30个肿瘤的外显子组序列数据。通过将这些结果与使用匹配的SNP阵列生成的结果进行比较,并通过肿瘤的等位基因特异性拷贝数分析(ASCAT)算法对其进行处理,从而评估这些算法的性能。
-
Sequenza / exome 和 SNP / ASCAT 之间的比较显示出细胞结构和多倍性估计值之间有很强的相关性。该性能明显优于以前发布的算法。 此外,在模拟正常肿瘤混合物的人工数据中,Sequenza在肿瘤含量低至30%的样品中检测到了正确的倍性。
Sequenza和基于SNP阵列的拷贝数图谱之间的一致性表明,单独进行外显子组测序不仅足以识别小规模突变,而且足以估计细胞数量和推断DNA拷贝数畸变。
一、介绍

Figure S1: Detailed schema of sequenza workflow Sequenza软件由两个不同的部分组成:
基于python的预处理工具 sequenza-utils
- 以FASTA格式从基因组参考文件计算滑动窗口中的GC含量;
- 处理来自肿瘤和正常标本的测序数据,这些数据为pileup格式由SAMtools输出;
- 脚本提取测序深度,确定正常标本中的纯合和杂合位置,并从肿瘤标本中计算出变异等位基因和等位基因频率。 输出为制表符分隔的文本文件,适合导入R。
sequenza R包,用于实现模型拟合和可视化功能
- sequenza.extract :有效地读取输入文件。对肿瘤进行GC含量归一化与正常深度之比,并使用“copynumber”软件包进行等位基因特异性分割。
- sequenza.fit :推断细胞性和倍性参数以及拷贝数分布图。 使用后验概率空间的局部最大值来提供替代解决方案。
- sequenza.results :返回估计结果以及替代解决方案以及沿基因组和单个染色体的数据和模型的可视化。
序列在肿瘤外显子测序数据中的应用
为了比较Sequenza WES图谱与当前最新的SNP图谱,从10例OVCA患者和20例KIRC患者中获得了配对的肿瘤正常外显子和Affymetrix SNP6阵列。之所以选择肾脏癌和卵巢癌,是因为它们代表了两种截然不同的癌症类型:透明细胞肾癌的细胞数低,拷贝数变异少,而卵巢癌通常表现出大量的拷贝数改变和高肿瘤细胞数。
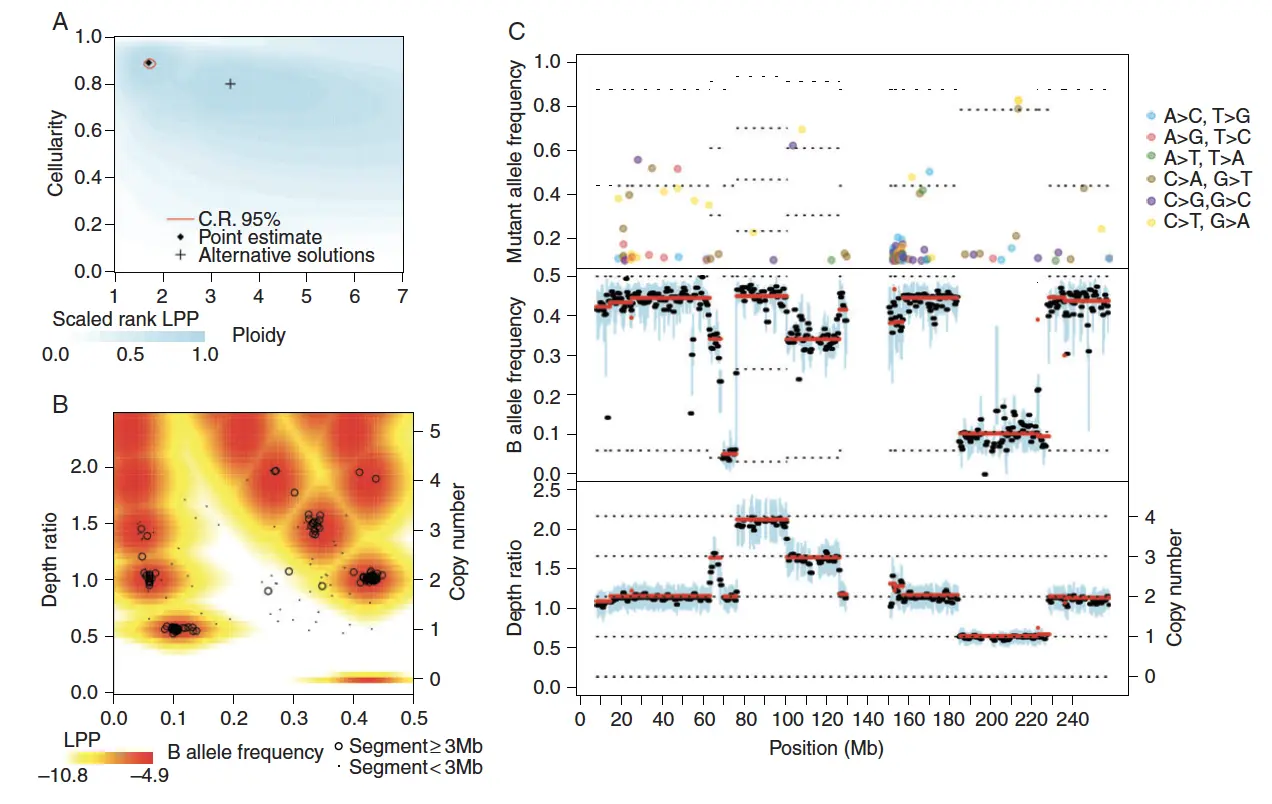
Figure 1. Representative output of the Sequenza algorithm
Figure 1.A 观察数据的对数后验概率(LPP)被计算为一系列候选倍性和细胞质数值
- Point estimate :具有最大LPP的倍性和细胞性。
- C.R.95% :95%置信区域是总后验概率大于0.95的最小(不一定是连续的)点集。
- 背景颜色:表示LPP的等级(蓝色=最可能,白色=最不可能)。
- Alternative solutions:局部最大值用“ + ”表示,并指出可能的替代解决方案。
Figure 1.B 观察每个基因组片段(黑色圆圈和点)的深度比和 BAF 值
- 代表性的联合LPP密度(颜色)是根据(A)中确定的细胞性和倍性估计值计算的,并且是根据假设的有代表性的10mb片段计算的。
- 实际连接 LPP 密度取决于节段大小和变异性,因此对于每个节段而言,其密度在数量上而非质量上有所不同。
- 观察到的部分极不可能的 DR 和 BAF 值可能表明亚克隆性,测量误差,或不正确的模型参数。 (555~其实这个图没看懂TT)
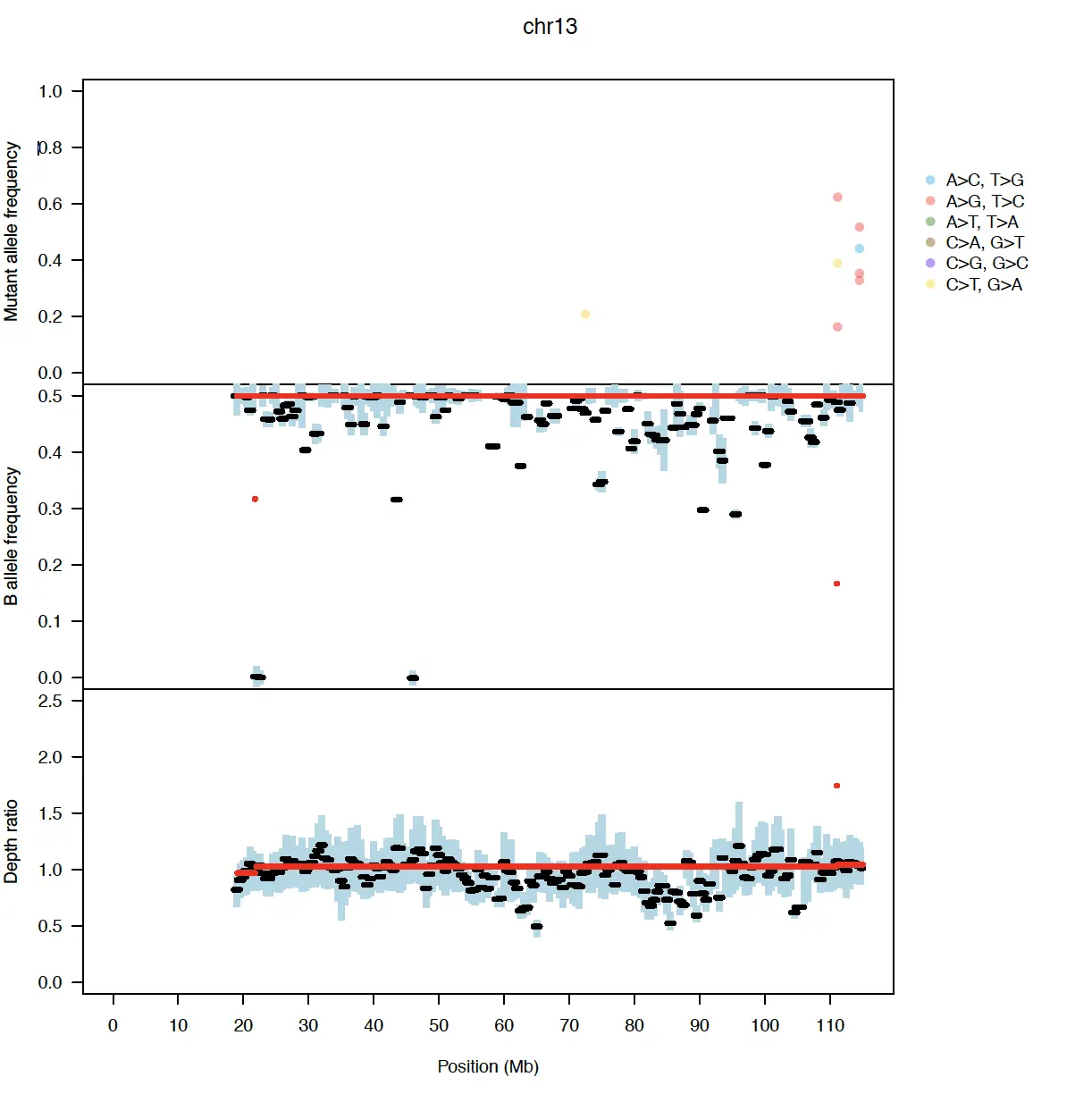
Figure 1.C 根据基因组位置绘制染色体图,指示MAF(上)、BAF(中)和DR(下)
- MAF:给定位置的MAF是具有突变的读数的分数,如果每个基因组位置> 0.1,且具有足够的测序深度,则显示该MAF。
- BAF、DR:将BAF和DR汇总在每0.5mb交错的1mb窗口内。每个窗口内粗黑线表示中值;蓝条表示四分位范围;红线表示分段的值;细虚线表示拟合模型下的期望值。
- 它们的位置分别基于估计的细胞数量、倍性和拷贝数分布。 在上图中,虚线表示具有突变的等位基因的数量,最低的线从1开始;在中图中,虚线表示次要等位基因拷贝数,最低的线从零开始;在下面的面板中,虚线表示copy number。
讨论
- Sequenza 与基于 SNP 数组的细胞质、倍性和拷贝数估计显示出更强的一致性。 然而,在细胞系外基因组数据的分析中,absCN-seq的表现优于Sequenza,这可能是Sequenza依赖于从匹配的正常样本中识别杂合子的位置,而这些样本对于这些细胞系是不可用的。
- Sequenza没有解释肿瘤标本中可能存在的突变异质性,这对患者诊断和驱动因子突变的鉴定有重要影响。
- 对于外部软件如 PyClone,可以使用不同的等位基因频率和相应的 Sequenza 的拷贝数状态作为输入,以解析子克隆结构。
二、软件
1. SAMtools输出pileup格式的肿瘤和正常标本数据。
samtools mpileup normal.sorted.bam -f genome.fa -Q 20 |gzip > normal.pileup.gz
samtools mpileup tumour.sorted.bam -f genome.fa -Q 20 |gzip > tumor.pileup.gz
2. python_sequenza-utils
#2.1 gc_wiggle生成全基因组GC文件
> python sequenza-utils.py GC-windows -w 50 genome.fa |gzip > hg38.gc50Base.txt.gz
#2.2 pileup2seqz生成合并seqz文件
> python sequenza-utils.py pileup2seqz -gc hg38.gc50Base.txt.gz -n normal.pileup.gz -t tumor.pileup.gz |gzip > out.seqz.gz
#2.3 seqz-binning修剪seqz文件
> python sequenza-utils.py seqz-binning -w 50 -s out.seqz.gz | gzip > out_small.seqz.gz
注意:也可用bam2seqz合并1和2.2步,直接从bam到合并seqz文件。
> python sequenza-utils.py bam2seqz -n normal.sorted.bam -t tumour.sorted.bam -f genome.fa -gc hg38.gc50Base.txt.gz -o > out.seqz.gz
3. R_sequenza_package
再次感叹一下安装R包真难!R-sequenza手册地址
> library(sequenza)
> seqz.data <- read.seqz("data.bin50_seqz.gz")
> head(seqz.data)
# A tibble: 6 x 14
chromosome position base.ref depth.normal depth.tumor depth.ratio Af Bf
<chr> <int> <chr> <int> <int> <dbl> <dbl> <dbl>
1 chrM 2 N 369 908 2.48 1 0
2 chrM 14 T 214 548 2.56 0.998 0
3 chrM 26 C 346 847 2.45 0.999 0
4 chrM 31 C 408 966 2.37 0.998 0
5 chrM 34 G 428 1042 2.44 0.999 0
6 chrM 43 C 586 1445 2.47 0.999 0
# ... with 6 more variables: zygosity.normal <chr>, GC.percent <dbl>,
# good.reads <dbl>, AB.normal <chr>, AB.tumor <chr>, tumor.strand <chr>
Normalize coverage by GC-content; Extract the information from the seqz file
> chromosome_list<-c("chr1","chr2","chr3","chr4","chr5","chr6","chr7","chr8","chr9","chr10","chr11","chr12","chr13","chr14","chr15","chr16","chr17","chr18","chr19","chr20","chr21","chr22")
> test <- sequenza.extract(data.bin50_seqz.gz,chromosome.list=chromosome_list)
> for(i in 1:22){
pdf(paste("figure2 Plot chromosome view chr",i,".pdf",sep=""))
chromosome.view(mut.tab = test$mutations[[i]], baf.windows = test$BAF[[i]],ratio.windows = test$ratio[[i]], min.N.ratio = 1,segments = test$segments[[i]], main = test$chromosomes[i])
dev.off()
}
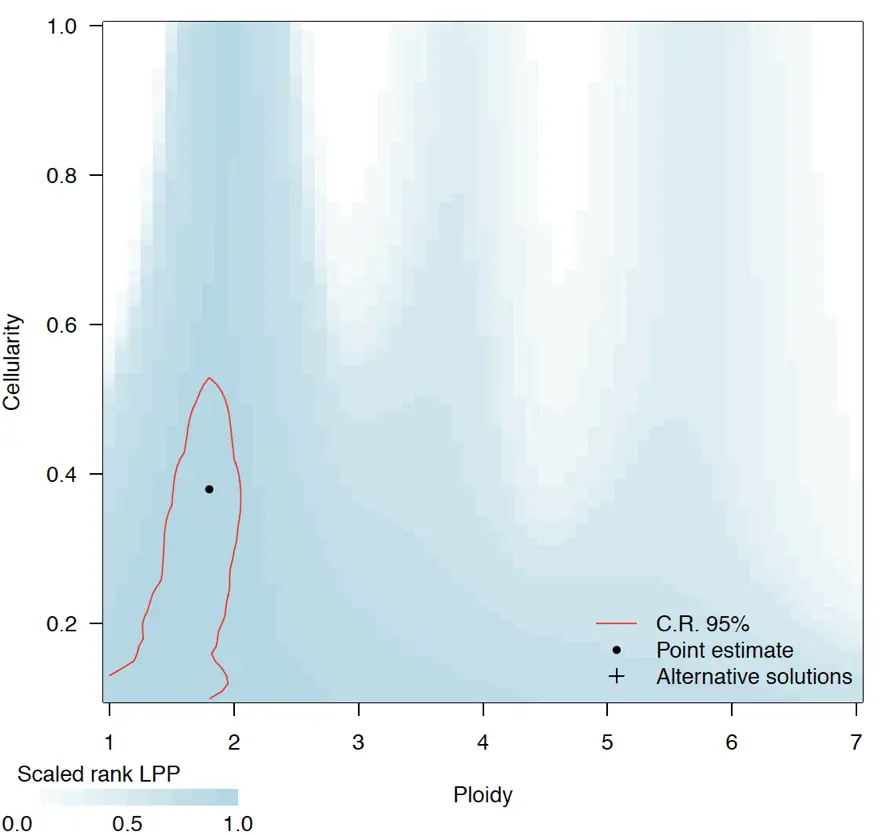
Inference of cellularity and ploidy
> CP.example <- sequenza.fit(test)
> pdf("figure2.5 cp.plot.pdf")
> cint <- get.ci(CP.example) #extract confidence intervals
> cp.plot(CP.example)
> cp.plot.contours(CP.example, add = TRUE, likThresh = c(0.95))
> dev.off()
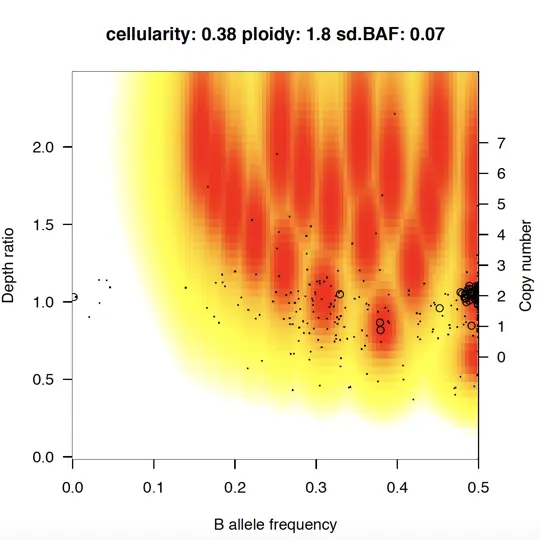
Results of model fitting
> sequenza.results(sequenza.extract = test, cp.table = CP.example,sample.id = "Test", out.dir="TEST")
sequenza.results后生成的TEST文件夹结果:
只能安慰自己一步步画可以对个别图修改特定格式,害!
> args<-commandArgs(T)
> library(sequenza)
> setwd("/out")
> seqz.data <- read.seqz("data.bin50_seqz.gz")
> #Inference of cellularity and ploidy
> CP.example <- sequenza.fit(result)
> #Results of model fitting
> sequenza.results(sequenza.extract = test, cp.table = CP.example,sample.id = "Test", out.dir="TEST")
参考资料
- https://www.jianshu.com/p/0889cc2adc63
- Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data 。 Annals of Oncology 26: 64–70, 2015 , doi:10.1093/annonc/mdu479 , Published online 15 October 2014
