【4.1.5】snpEff对基因型VCF文件进行变异注释
snpEff是一个用于对基因组单核苷酸多态性(SNP)进行注释的软件,snpEff软件可以用于对VCF文件进行变异注释,使用时需要先进行安装,然后构建参考基因组数据库,即可对VCF文件进行注释,下面进行用法介绍。
一、安装
首先安装好java环境,通过官网下载最新版本的软件压缩包,然后解压即可,最好安装在自己熟悉的目录下。
# Download latest version
wget https://snpeff.blob.core.windows.net/versions/snpEff_latest_core.zip
# Unzip file
unzip snpEff_latest_core.zip
另外,推荐一种更加简单快捷的方法,直接使用conda安装,命令如下:
conda install snpeff -c bioconda
配置数据库
有一些物种已经有官方的注释数据库,可以直接进行下载, 比如人类基因组,用如下代码下载:
java -jar snpEff.jar download GRCh38.76
如果需要查找哪些物种有现成的数据库,可以使用如下命令:
java -jar snpEff.jar databases
如果官方没有给出数据库,就需要自行建立数据库,以下用小麦举例,通过参考基因组和注释文件建立注释数据库。
下载数据文件
需要两个主要文件,iwgsc_refseqv2.1_assembly.fa是组装好的基因组序列,iwgscRefseqv2.1HCLC.gff3是注释文件,这两个文件可以在 https://wheatgenome.org/Projects/Reference-Genome-Project/RefSeq-v2.1 下载,如果是研究其他物种,也可以在网络上找到这两个文件。
配置文件修改
需要修改的配置文件在snpEff目录下, 文件名为snpEff.config,打开这个文件,在最后一行添加自定义数据库信息(这里的wheat可以自定义,但是要保持一致)
echo "wheat.genome:wheat" >> snpEff.conf
数据库路径设置
按照如下设置进入snpEff的安装目录并创建文件夹:
cd snpEff #进入 snpEff 目录下
mkdir data #新建 data 目录
cd data #进入 data 目录下,必须在该目录
mkdir genomes #新建 genomes 目录,用于建立 wheat 目录中的 bin
mkdir wheat #新建 wheat 目录,对应物种名或后续软件调用的参数名
将参考基因组.fa文件放在genomes文件夹中,并改名为wheat.fa,并将gff注释文件放在wheat文件夹中改名wheat.gff,最终形成如下文件结构。
$ tree
.
├── genomes
│ └── wheat.fa -> /NGS/Ref/IWGSC_V2.1_fa_gff/iwgsc_refseqv2.1_assembly.fa
└── wheat
├── genes.gff -> /NGS/Ref/IWGSC_V2.1_fa_gff/iwgscRefseqv2.1HCLC.gff3
运行脚本形成bin文件
准备好文件后,回到软件目录,并执行以下命令,自动生成参考文件。检查一下存放参考基因组注释文件的目录下是否出现一些以.bin结尾的文件(数量与参考基因组染色体数有关),有就代表构建成功。
java -jar snpEff.jar build -gff3 -v wheat -d -noCheckCds -noCheckProtein
├── sequence.1A.bin
├── sequence.1B.bin
├── sequence.1D.bin
├── sequence.2A.bin
├── sequence.2B.bin
├── sequence.2D.bin
├── sequence.3A.bin
├── sequence.3B.bin
├── sequence.3D.bin
├── sequence.4A.bin
├── sequence.4B.bin
├── sequence.4D.bin
├── sequence.5A.bin
├── sequence.5B.bin
├── sequence.5D.bin
├── sequence.6A.bin
├── sequence.6B.bin
├── sequence.6D.bin
├── sequence.7A.bin
├── sequence.7B.bin
├── sequence.7D.bin
├── sequence.Unknown.bin
└── snpEffectPredictor.bin
数据库构建完成后就可以直接用了,下一次不用再重新弄。
二、使用方法
2.1 准备内容
- 环境:Linux 或 Ubuntu,已经安装openjdk
- 文件:基因型变异信息 VCF 格式的文件
- 参考文件:gff 或 gtf 注释文件、参考基因文件
- 软件:SnpEff
2.2 运行程序
如果是通过conda安装,直接运行以下命令即可调用,对vcf文件进行注释。
snpEff wheat ./xxx.vcf.gz > ./xxx_snpeff.vcf.gz
如果是通过本地安装,可以用java调用程序进行计算,推荐使用这种方法,更加稳定。
java -jar ~/snpeff-5.1-2/snpEff.jar
-c ~/snpeff-5.1-2/snpEff.config wheat
../xxx.vcf.gz > ./xxxsnp.vcf.gz
等待注释完成后会生成snpEff_genes.txt文件和snpEff_summary.html文件,记录了注释的摘要信息,另外生成一个新的vcf文件包含详细注释信息。
2.3 结果查看
运行完成后会生成一个html的网页文件,里面记录了很多重要信息,接下来进行解读(参考知乎大佬天火三玄变的帖子)
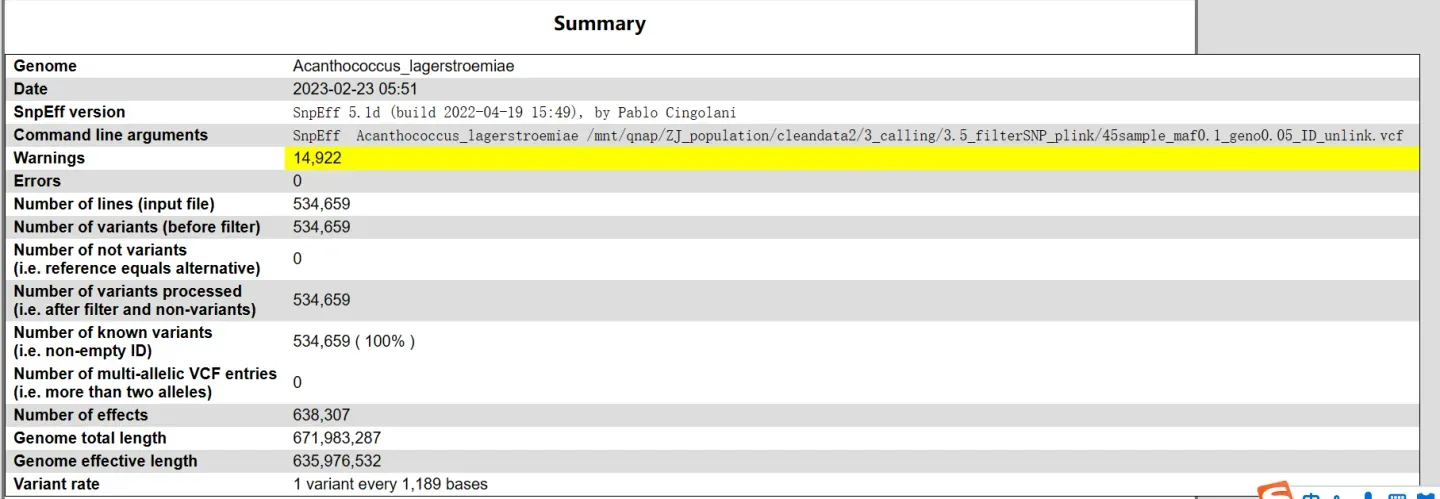
摘要信息
从上往下依次是:基因组(物种名)、注释日期、注释命令、警告信息、错误信息、输入文件行数、变异位点数(过滤之前)、非变异位点数(与参考基因组碱基一致)、变异位点数(过滤之后)、具有ID的变异位点数、非双等位基因组SNP位点数、effects个数、参考基因组总长度、参考基因组有效长度、变异率(参考基因组有效长度/变异位点数)
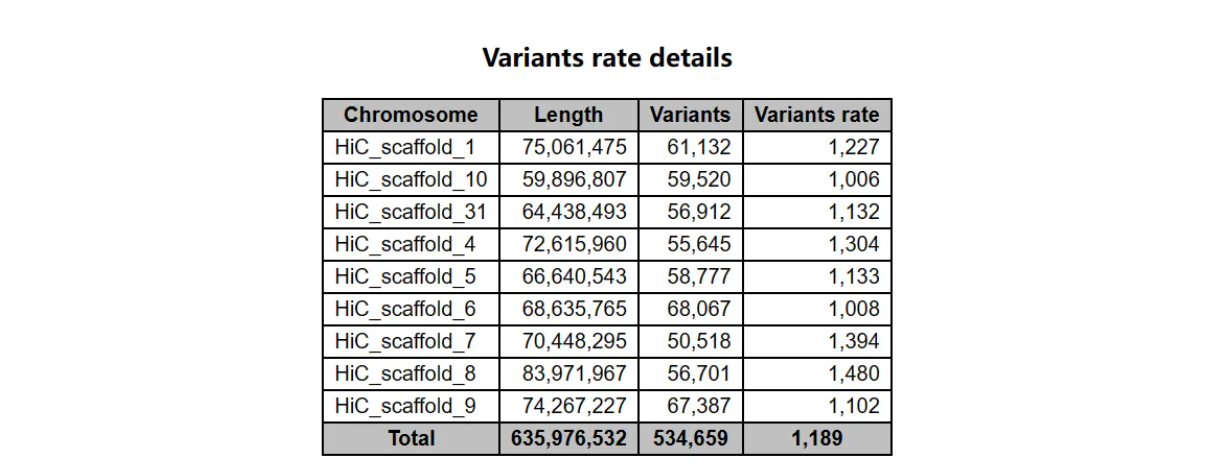
各染色体变异率
从左往右:染色体编号、长度、变异位点数、变异率(多少个碱基中有一个变异位点)
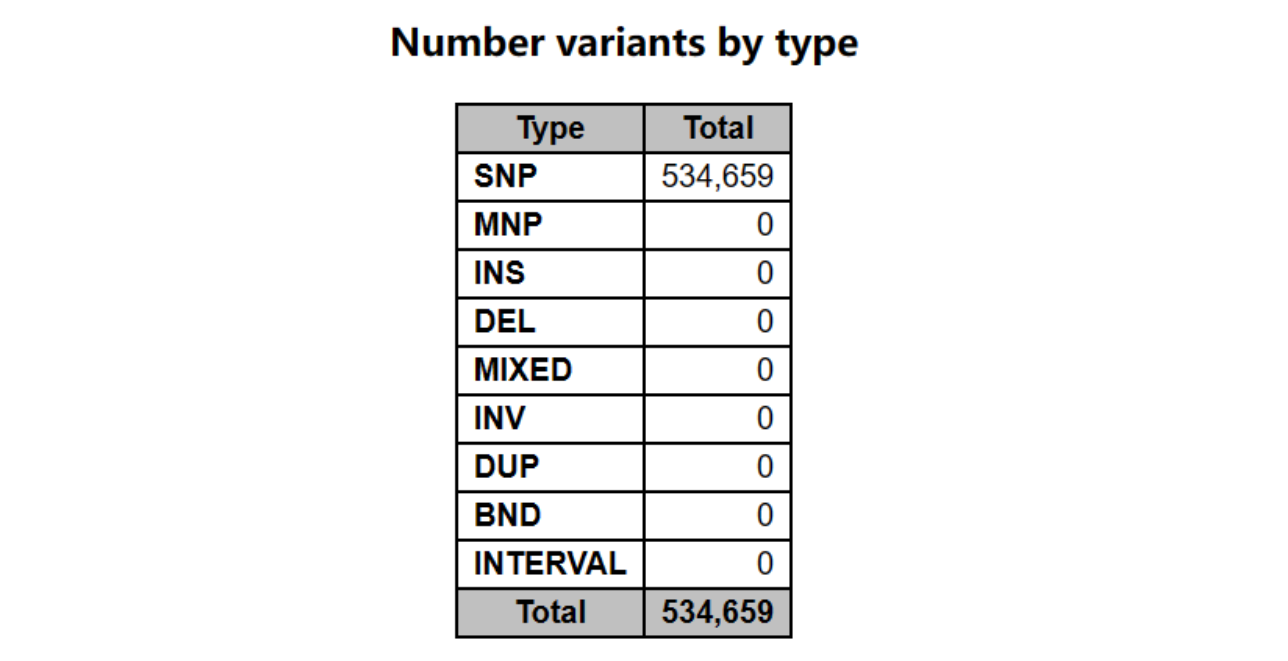
变异类型
包括:SNP(单核苷酸多态性)、MNP(多核苷酸多态性)、INS(插入变异)、DEL(缺失变异)、MIXED(混合变异)、INV(倒位变异)、DUP(重复变异)、BED(易位变异)、INTERVAL(间隔变异)
有效影响数量

功能分级有效数: MiSSENSE(错义突变)、NONSENSE(无义突变)、SILENT(沉默突变)

有效变异数和百分比
下图左边为按类型划分有效变异数,包括(从上往下):3’端主要UTR变异(UTR是成熟mRNA分子5’或3’端不被翻译的部分,一般在mRNA转运、稳定性和翻译调节中起重要作用)、5’端主要UTR提前启动子获得变异、5’端主要UTR变异、下游基因变异、起始密码子编码变异、基因间隔区、内含子变异、剪接受体变异、剪接供体变异、剪接区域变异、起始缺失、起始保留变异、终止获得、终止缺失、终止保留变异、同义变异、上游基因变异。
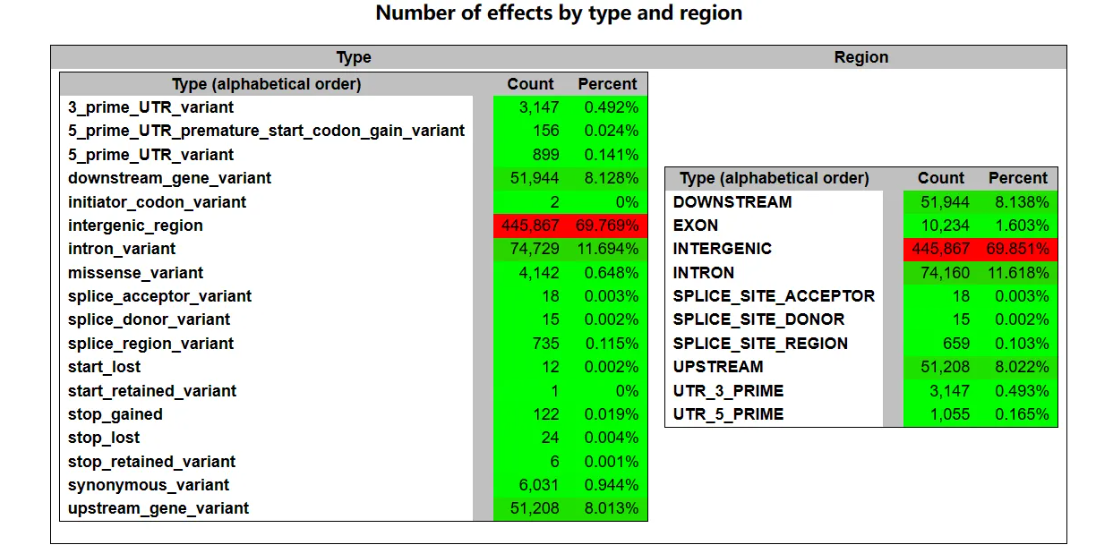
右边为按区域划分有效变异数,包括(从上往下):下游、外显子、间隔区、内含子、剪接位点受体、剪接位点供体、剪接位点区域、上游、3’UTR区、5’UTR区。
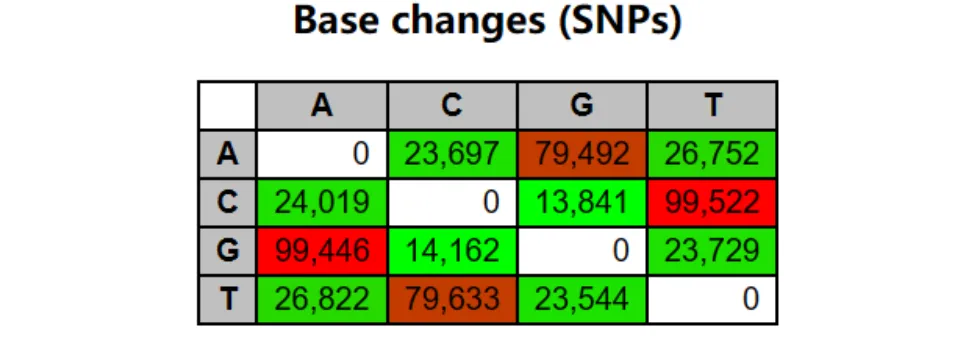
SNP位点碱基变异表
可以看出SNP中哪些碱基的转换比较多(A腺嘌呤、C胞嘧啶、G鸟嘌呤、T胸腺嘧啶)
总结:
在使用snpEff过程中需要注意数据库的选择和构建,根据不同版本进行计算,另外尽量避免更改染色体的展示方式,防止造成识别错误。另外可以利用vcftools将vcf中的样品信息去掉,这样文件体积会大大缩小,有利用加快注释速度。
2.4 结果说明
https://blog.51cto.com/u_10721944/5398047
三、我得案例
export PATH=/opt/bin/jdk-12.0.2/bin:$PATH
java -jar /opt/bin/snpEff/snpEff.jar -c /opt/bin/snpEff/snpEff.config -formatEff -classic -noStats -noLog -quiet -no-upstream -no-downstream -v sars-cov-2 ${output}/${ID}/${ID}.vcf > ${output}/${ID}/${ID}.ann.vcf
参考资料
