【2.1.1】Somatic与Germline 变异检测
一、Somatic与Germline 变异检测背景
以下用表格整理两种变异形式在形成原因、遗传性的方面等特点
| null | 一般解释 | 形成原因 | 遗传性 |
|---|---|---|---|
| Germline Mutation | 胚系突变,胚系突变可以遗传给后代,子代的所有细胞均应带有一致的胚系突变,但不一定致病 | 生殖细胞(germ cells)突变,女性中为卵细胞,男性中为精源细胞 | 有概率遗传给子代 |
| Somatic Mutation | 体细胞突变,不能够遗传给后代 | 体细胞(somatic cells)突变,除升职细胞外,例如皮肤,肝脏,骨髓,眼睛等的细胞,1.某些致癌物质,如紫外线、辐射、化学暴露;2.原癌基因或抑癌基因发生突变;3.病毒感染 | 由于非生殖细胞突变,一般情况下生物个体死亡改突变停止继承(特殊情况如海拉细胞,植物的无性繁殖与扦插) |
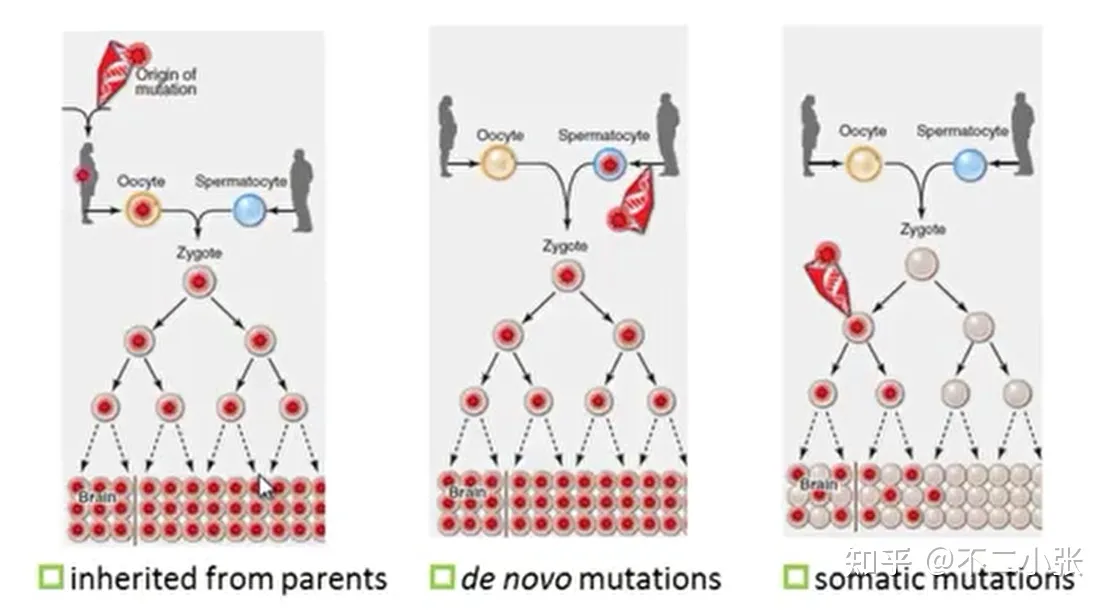
另外,这两种类型的变异在发生时期也存在差异:
Germline mutations发生在「减数分裂」的过程中,因为性细胞需要将其染色体分裂成两半,以便在受精过程中恢复原始的染色体数。 在精子中发生这种突变的可能性要比在卵子中发生的可能性高,因为前者形成过程中发生的细胞分裂数量要多于后者,这使得雄性的种系突变率更高。
Somatic mutations发生在「有丝分裂」期间,所有体细胞仅通过该过程分裂。 这是因为,有丝分裂产生的子细胞具有相等数目的染色体,这对于维持细胞中的人类染色体数目至关重要
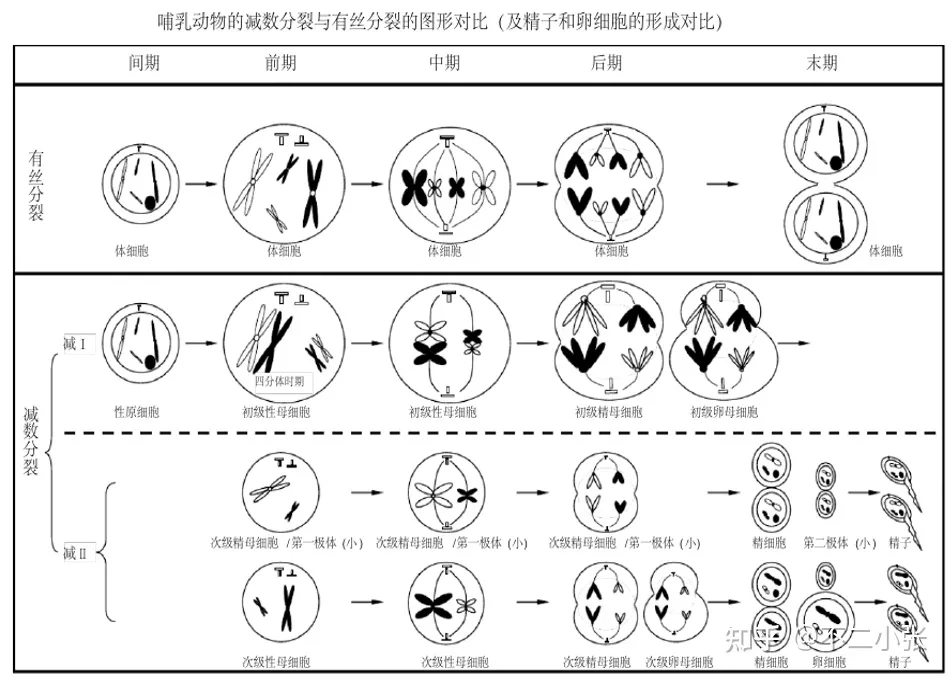
人类基因组为二倍体,意味着同样一个位点的检测,基因型可能是纯合,也可能是杂合的。 对于两类示例的临床案例如下:
1.Germline mutations
-
突变的概率一般是:[0, 0.5, 1],双亲均突变,则是1,其中一方突变是0.5,都没有突变就是0;
-
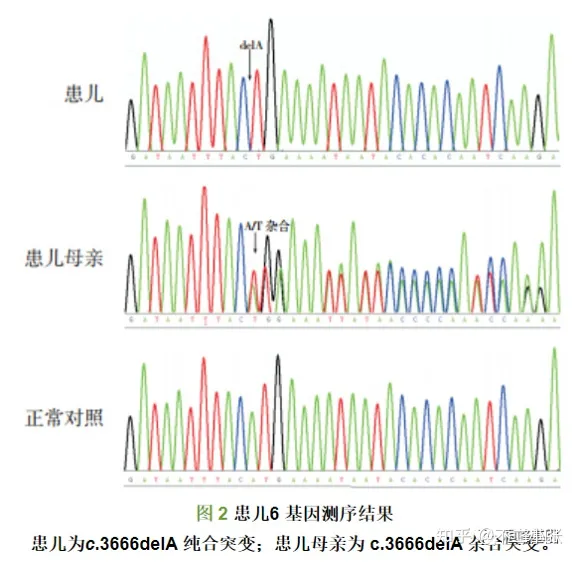
临床案例:血友病A(Hemophilia A,HA)为X 连锁隐性遗传性凝血功能障碍性疾病,患病率为1/5 000~10 000。 患儿6 检出基因型p.His1202LeufsX16(c.3666delA),其母亲为杂合子,该基因型在HAMSTeRS、人类基因突变数据库以及美国国立生物技术信息中心等数据库中均未能检索到,50 例正常男性中亦未检测到相同基因变异,NCBI 数据库未检索到多态性基因记录,提示为新突变类型。
2. Somatic mutations
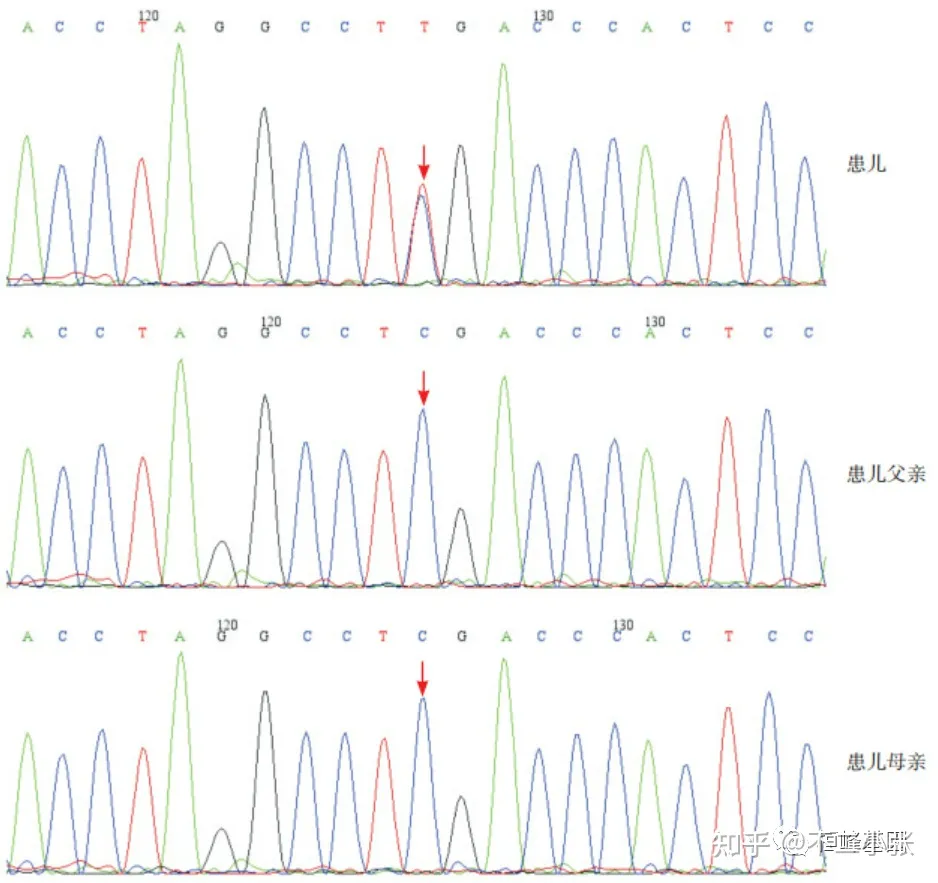
- 临床案例:Floating-Harbor综合征(FHS),由SRCAP基因突变引起的常染色体显性遗传病,患儿表现为特殊面部特征(三角脸、嘴唇薄、长睫毛)、骨骼畸形(手指弯曲)、语言表达障碍、骨龄落后,基因检测显示SRCAP基因存在新发c.7330C > T (p.R2444X)杂合突变。
3. Somatic与Germline Mutations 与遗传性疾病或肿瘤分析的关系
- Somatic mutations 一定不是导致遗传性疾病的突变(密码子简并性,中心法则下编码的蛋白质的氨基酸序列未必发生改变)
- Somatic mutations 与 Germline Mutations 的发生,不一定致病
- Germline Mutations 是遗传性疾病的发病原因,但和环境的相互作用也同样存在关系
- Somatic mutations 不一定是直接导致肿瘤形成的直接机制,Germline Mutations 可能与肿瘤的形成、发展也有相关性
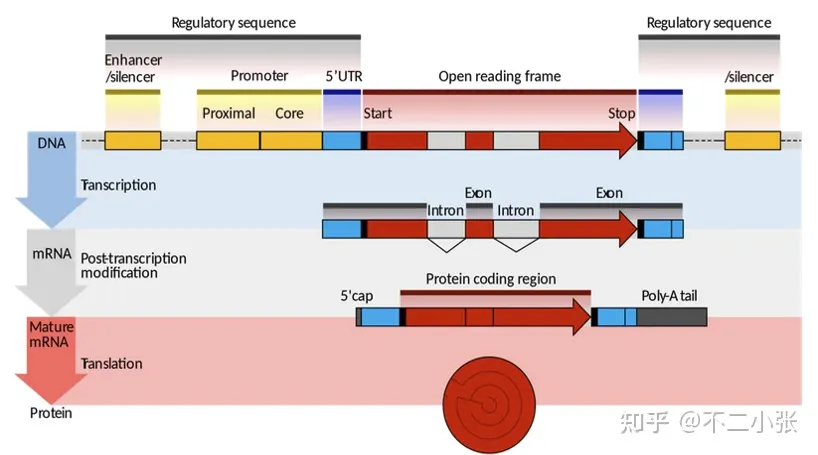
二、一般分析策略
2.1 分析策略
Germline突变频率主要集中在50%(杂合) , 100%(纯合),一般是比对错误或者测序错误等原因产出噪音,干扰变异探测。我们可以使用统计学分析或者机器学习等方法,对潜在变异位点进行区分,得到突变位点。 Somatic一般使用对照样本进行分析, 例如,取肿瘤细胞作为Tumor样本,取血液中的白细胞或者癌旁正常细胞作为对照样本来分析,Tumor中特有的突变为Somatic mutation

基于不同的突变类型,变异calling采用不同的软件,大体上突变类型如下:
2.2 SNPs:置换、颠换
snp和indel一般在一般软件中,可以同时进行calling,常见的软件为GATK
Germline Mutation calling(将样品基因组序列与参考样本(父母本)与参考基因组序列进行比对)
$(gatk) --java-options "-Xmx6g" HaplotypeCaller -ERC GVCF \
-R $(fasta) \
-I $(sample_id).bam \
-L $(Agilent_64M_bed) \
-O $(sample_id)_HaplotypeCaller.g.vcf
$(gatk) --java-options "-Xmx20g" CombineGVCFs \
-R $(fasta) \
-V $(child)_HaplotypeCaller.g.vcf \
-V $(father)_HaplotypeCaller.g.vcf \
-V $(mother)_HaplotypeCaller.g.vcf \
-L $(Agilent_64M_bed) \
--dbsnp $(dbsnp_vcf) \
-O $(trio)_HaplotypeCaller.raw.g.vcf
$(gatk) --java-options "-Xmx20g" GenotypeGVCFs \
-R $(fasta) \
-V $(trio)_HaplotypeCaller.raw.g.vcf \
-L $(Agilent_64M_bed) \
--dbsnp $(dbsnp_vcf) \
-O $(trio)_GenotypeGVCFs.vcf
Somatic Mutation(即将样品基因组序列与参考样本(癌旁组织)与参考基因组序列进行比对),一般检测方法如下
- 通过计算等位基因(counting alleles)
- 通过二项分布( binomial distribution)
- 通过泊松二项分布(Poisson-binomial distribution)
- 通过贝叶斯方法(Bayesian)
运行:
gatk --java-options "-Xmx32g" Mutect2 \
-R fasta \
-I tumorBAM -tumor samplenameA \
-I normalBAM -normal samplenameB \
-O gatk4.vcf.gz
gatk --java-options "-Xmx8g" FilterMutectCalls \
-R fasta \
-V gatk4.vcf.gz \
-O gatk4.raw.vcf.gz \
-XL wgEncodeDacMapabilityConsensusExcludablehg38.bed
2.3 InDel:小的插入或缺失(<50bp)

CNV 拷贝数变异:狭义上指拷贝数的重复、丢失,大小介于 1kb 至 3MB 的 DNA 片段的变异,基因的计量效应-ACMG
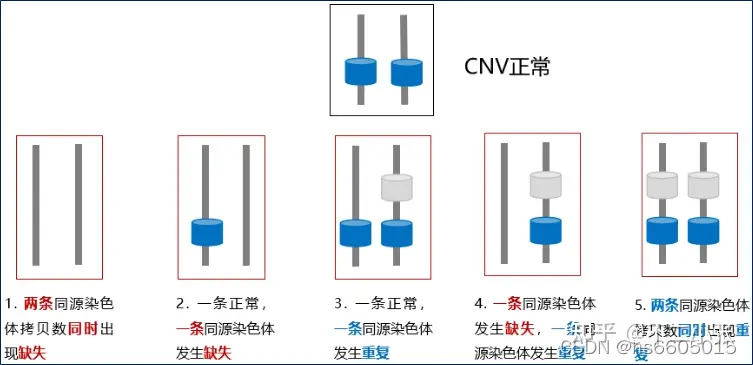
Germline Mutation:即将样品基因组序列与参考样本(父母本)与参考基因组序列进行比对,通过计算两者之间的拷贝数比率判断CNV的存在。 覆盖基因组范围的3.7% to 12%,不同人群中重叠程度较高。软件中一般采用cnvkit进行calling,核心家系样本CNV calling参考使用方式如下:
#!/bin/bash
# --ploidy 选项来指定探测样本的染色体倍性为 2,--ploidy-caller 选项来指定染色体倍性估计方法为 clonal,--ploidy-clusters 选项来指定染色体倍性聚类文件,--annotate 选项来注释 CNV
# install CNVkit via conda
conda install -c bioconda cnvkit
# set up environment
source activate cnvkit
# define input and output files
input_paternal_bam="/path/to/paternal.bam"
input_maternal_bam="/path/to/maternal.bam"
input_proband_bam="/path/to/proband.bam"
output_paternal_cnr="/path/to/paternal.cnr"
output_paternal_cns="/path/to/paternal.cns"
output_maternal_cnr="/path/to/maternal.cnr"
output_maternal_cns="/path/to/maternal.cns"
output_proband_cnr="/path/to/proband.cnr"
output_proband_cns="/path/to/proband.cns"
output_calls="/path/to/calls.txt"
output_scatter="/path/to/scatter.png"
# run CNVkit batch for each sample
cnvkit.py batch $input_paternal_bam -o $output_paternal_cnr --output-reference $output_paternal_cns
cnvkit.py batch $input_maternal_bam -o $output_maternal_cnr --output-reference $output_maternal_cns
cnvkit.py batch $input_proband_bam -o $output_proband_cnr --output-reference $output_proband_cns --father $output_paternal_cnr --mother $output_maternal_cnr
# call CNVs for proband using parents as reference
cnvkit.py call $output_proband_cns -o $output_calls --ploidy 2 --ploidy-caller clonal --ploidy-clusters $output_proband_cnr.cluster.txt --annotate $output_paternal_cns,$output_maternal_cns
# generate scatter plot
cnvkit.py scatter $output_calls -s $output_scatter
cnvkit.py diagram $output_calls -s $output_cns.diagram.png
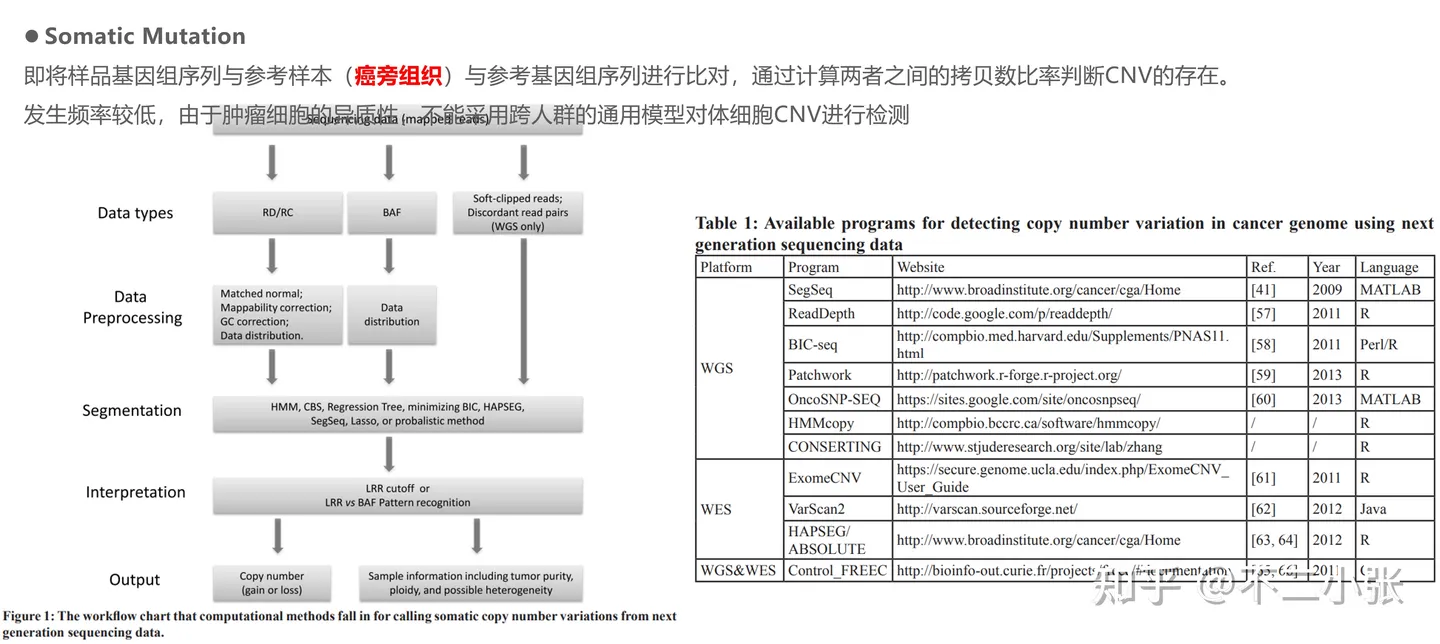
Somatic Mutation:即将样品基因组序列与参考样本(癌旁组织)与参考基因组序列进行比对,通过计算两者之间的拷贝数比率判断CNV的存在。 发生频率较低。
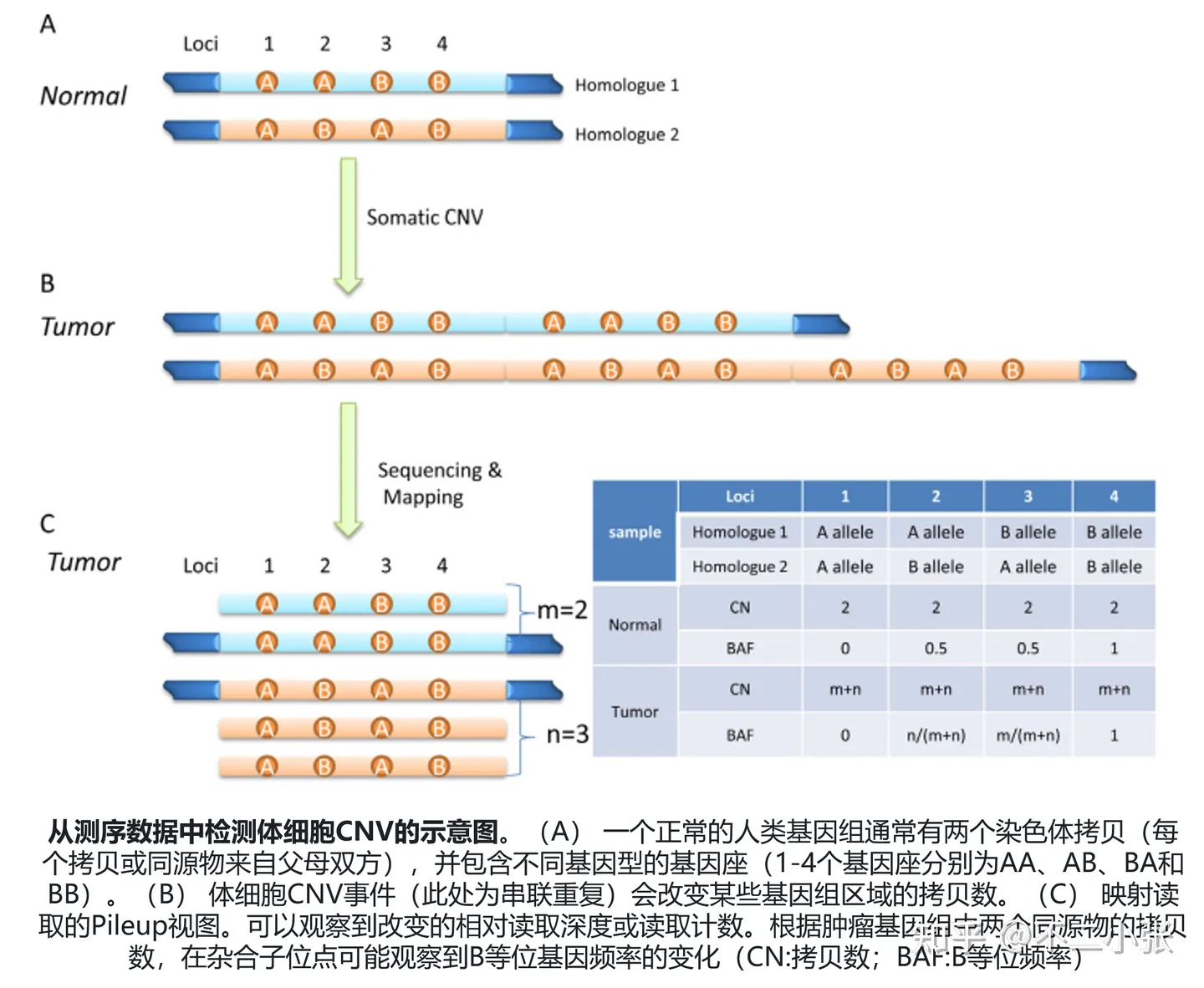
在CNV有效性检测中,有一些软件采用BAF(B-Allele Frequency)作为指标,。当存在存在CNV,那么在该位点上被检测到的拷贝数会与该个体其他位点上的拷贝数不一致,需要计算该位点上两种碱基的频率。对于二倍体来说,如果一个个体没有CNV,那么该位点上两种碱基的频率应该接近0.5,因为每个染色体上该位置上的两个碱基应该是等量的。然而,如果一个个体某一染色体上发生了CNV,则该位点上两种碱基的频率可能会大幅偏离0.5。因此,BAF值就是该位点上较少的碱基类型的频率。需要注意的是,BAF值只能用于二倍体的样本,对于多倍体的样本无法使用。此外,在计算BAF值时也需要考虑一些影响因素,例如测序深度和测序错误率等。
由于肿瘤细胞的异质性,不能采用跨人群的通用模型对体细胞CNV进行检测。下图介绍了常用的Somatic CNV calling的工具和它实现的算法,以其中的Control_FREEC为例进行说明。 配置config.txt文件
[general]
[general]
chrFiles = ./Chr/
chrLenFile = hg38_chrLength.no_chrM.txt
ploidy = 2
window = 0
breakPointType = 4
maxThreads = 6
breakPointThreshold = 0.8
BedGraphOutput = TRUE
outputDir = .
assumeUniqueMap = 1
maxThreads = 4
telocentromeric = /path/to/hg19_telocentromeric.txt
[sample]
mateFile = tumor.bam
inputFormat = BAM
mateOrientation = FR
[sample]
mateFile = normal.bam
inputFormat = BAM
mateOrientation = FR
[BAF]
SNPfile = ./dbSNP147_hg38.vcf
minimalCoveragePerPosition = 5
shiftInQuality = 33
[target]
captureRegions = Agilent_64M.bed
运行代码如下:
# 安装 Control-FREEC
conda install -c bioconda control-freec
# 准备输入文件
# 将正常和肿瘤样本的 BAM 文件列表保存到一个文本文件中,例如 tumor_normal_bamlist.txt
freec -conf config.txt
# 使用自带的绘图工具生成结果
# 以下示例命令假设 Control-FREEC 的输出文件为 sample_CNVs.txt,结果图保存为 sample_CNVs.png
freec_plot.pl -conf config.txt -output sample_CNVs.png -png -title "Sample CNVs" sample_CNVs.txt
在肿瘤CNV calling中,许多因素都会影响最终的效果,如下
-
倍性: 指肿瘤细胞所含染色体组数的变化情况,常见的倍性有二倍体、三倍体、四倍体等。在肿瘤分析中,如果肿瘤样本的倍性发生变化,则会对CNV(基因拷贝数变异)分析产生极大影响。比如,对于一个拷贝数变异的区域,在二倍体肿瘤中可能被检测到,但在四倍体肿瘤中则可能无法被检测到。因此,倍性变化会导致肿瘤分析的灵敏度和特异性发生变化,需要根据具体情况进行调整和优化。
-
纯度: 指肿瘤样本中包含肿瘤细胞的百分比。在肿瘤样本中,存在正常组织和肿瘤组织的混合情况。当纯度较高时(例如90%以上),容易检测到更低频率的变异位点,因为该变异可能出现在大多数肿瘤细胞中。而当纯度较低时(例如50%以下),可能会忽略低频率或个别的变异位点,基因组非整倍性情况的存在会严重影响肿瘤样本中BAF的状态,以及read count和read depth的基线。
-
克隆结构异质性
-
亚克隆: 体现了肿瘤的异质性,亚克隆对肿瘤进化和癌症复发很重要。由于它们在样本中的百分比较低,因此很难确定亚克隆,不过增加测序深度有助于捕获大量亚克隆。
结构变异,又分为具体的一下几种类型

- 插入 (Insertion)
- 缺失 (Deletion)
- 反转 (Inversion)
- 染色体内易位 (Intra-chromosomal Translocation)
- 染色体间易位 (Inter-chromosomal Translocation)
- 拷贝数变异 (Copy Number Variation)
- 一些形式更为复杂的变异
由于SV是一个非常复杂的结构变异场景,每一种的细节处理方式不一定相同,详细的信息可以关注最近进展的文献。
参考资料
