【2.3.5】推断结构变异(SV)断点的癌细胞分数(ccf)
Inferring structural variant cancer cell fraction推断结构变异癌细胞分数
一. 研究背景
癌症进化的克隆理论认为,癌症源自单个祖细胞,该祖细胞已获得赋予选择性优势的突变,从而导致了遗传上相同的细胞群体或克隆的扩展。随着癌症的发展,出现了类似于达尔文物种进化的过程,随后通过不断获取优势基因组畸变从始发克隆(founding clone)中产生了随后的遗传上不同的种群。因此,肿瘤很可能由多个细胞群体的遗传异质组合组成,其程度已通过使用全基因组测序得以揭示。
对此,作者提出了SVclone,这是一种从全基因组测序数据推断结构变异(SV)断点的癌细胞分数(cancer cell fraction,CCF)的计算方法,包括拷贝数异常和拷贝数中性变异。作者使用来自同一患者的两次克隆转移的已知比例的真实样品的计算机模拟混合物评估性能。作为全基因组全癌症分析(PCAWG)联盟的一部分,该联盟汇总了38种肿瘤类型中2658例癌症的全基因组测序数据,作者使用SVclone揭示了肝癌,卵巢癌和胰腺癌的一个子集,其中亚克隆丰富的拷贝数中性重排(copy- number neutral rearrangements)表明总体癌细胞存活率下降。SVclone可使SV肿瘤内异质性的表征更明显。
癌细胞分数(CCF):如果肿瘤起源于一个单个细胞(癌症进化的克隆理论认为,癌症源自单个祖细胞),该肿瘤被认为是一个克隆,起始突变存在于每一个肿瘤细胞中,被称为CCF为1,即癌细胞是克隆性的,由普遍扩散所致。CCF<1的细胞组成的肿瘤称为亚克隆,即正在进行的扩增的一部分。但是,事实情况下即便一个特定的突变出现在一次活检中,CCF为1,在接下来的肿瘤采样中可能部分或者完全检测不到这个突变,因为观察到的变异等位基因频率取决于正常细胞混合物(纯度)的数量和局部拷贝数。
肿瘤异质性:肿瘤的异质性是恶性肿瘤的特征之一,是指肿瘤在生长过程中,经过多次分裂增殖,其子细胞呈现出分子生物学或基因方面的改变,从而使肿瘤的生长速度、侵袭能力、对药物的敏感性、预后等各方面产生差异。
二. 分析流程
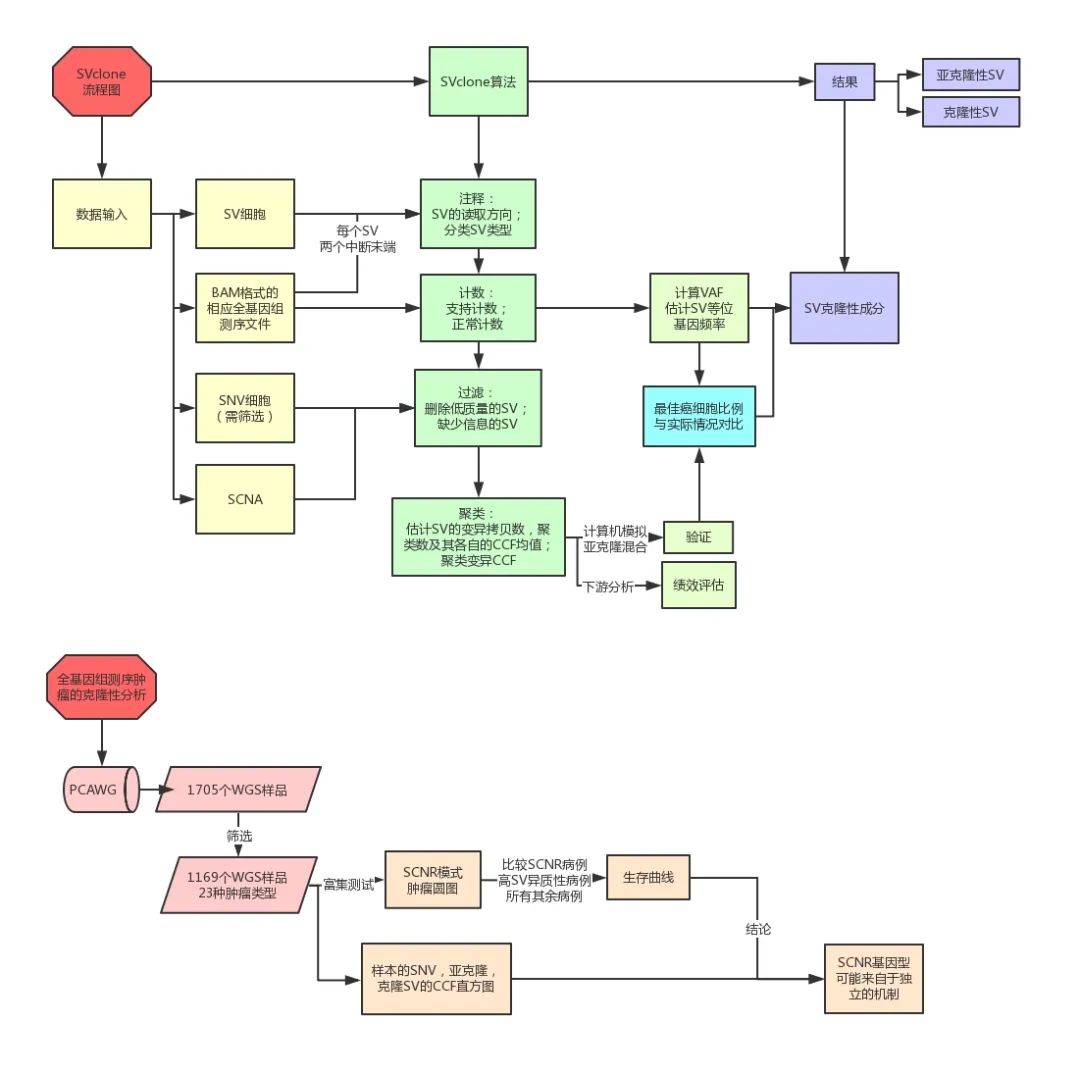
三. 结果解读
1. 算法描述
SVclone算法包含五个步骤:注释,计数,过滤,聚类和后续分配(post-assign)。
- 注释:SV调用是注释步骤(单核苷酸分辨率配对的SV位点)以及BAM格式的相应全基因组测序文件的输入。带注释的步骤确定了SV的读取方向,并分类了SV类型;
- 计数:计数步骤估计支持和正常(normal,不支持)读取计数,并计算SV VAF。
- 过滤:删除低质量的SV和缺少信息的SV,并在给定拷贝数调用的情况下,推断每个中断末端(break-end)的背景拷贝数(background copy number)。
- 聚类:聚类步骤同时估计SV的变异拷贝数,聚类数及其各自的CCF均值。来自每个SV的两个末端的等位基因频率用于进行推断。
- 后续分配:给定先前获得的聚类配置,后(重新)分配步骤为变异分配最可能的变异拷贝数和CCF。
VAF:突变reads占总reads的比例(variant allele fraction)
2. SV等位基因频率的估计
SV等位基因频率可以用与SNV相同的方式估算:变异读段数除以SV断点处观察到的读段总数。SV面临的挑战是,许多读取跨断点被拆分,从而难以对这些读取计数提取准确的估计。作者在不同的肿瘤纯度下模拟了具有已知等位基因频率的SV的读数,之后实现了一种优化方法用于根据这些读取计数来计算VAF。模拟结果表明,VAF估计值是准确的,与纯度无关,但重复项除外(图1c)。其中右图表示,其中以20-100%的纯度水平以20%的增量递增,其中预期的VAF是纯度的一半(虚线)。
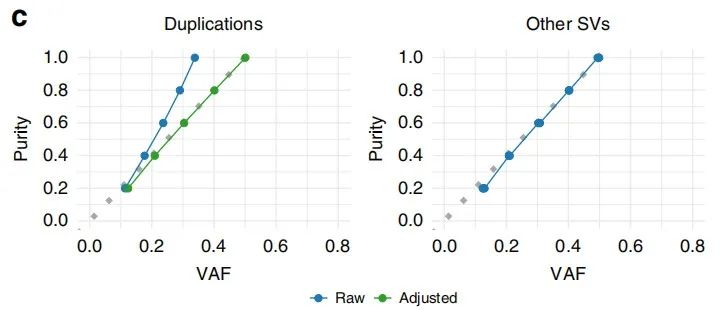
图1c. 重复调整原始VAF的效果(左),其他SV调整未调整的VAF的效果(右)
由于DNA获取(DNA-gains)的增加,复制显示正常读取计数增加,而正常DNA没有丢失(图1b)。其中,从左到右,每个片段显示一个不受影响的基因座,变异类型对断点处读取的影响以及标准化等位基因频率所需的调节策略。读取的红色部分显示软切换(soft-clips),即读取的部分映射到断点(breakpoint)的另一端。
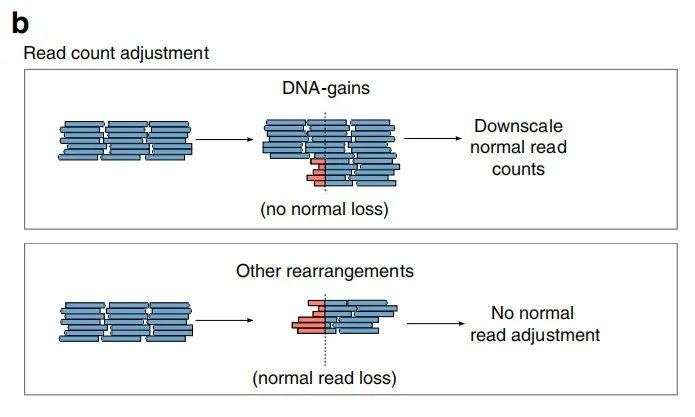
图1b. DNA获取(顶部)和所有其他重排(底部)所需的调整
为了解决这一偏见,作者引入了一个尺度因子(scaling factor),该比例因数可整合肿瘤纯度以校准支持的读取计数。这可以纠正偏差,并显示出对潜在VAF(underlying VAF)的准确估计(图1c左)。
3. 在计算机上对肿瘤进行亚克隆混合以进行验证
目前的评估推断CCF突变的算法的性能仅限于模拟SNV和拷贝数更改,还没有用于测试性能SV癌细胞分数推断的黄金标准数据集。因此,作者创建了具有已知SV亚克隆结构的肿瘤样品的数据集。通过计算机模拟,作者选择以已知的亚克隆比例混合来自同一患者的两个全基因组测序样品(图2a)。通过混合肿瘤序列数据,作者保持了真实序列数据的许多噪声特征(noise characteristics)。样本包括一组三聚类混合物,其中SV和SNV以已知的克隆频率以10%的增量(increments)进行二次采样,以及通过对不同频率的奇数和偶数染色体进行二次采样而创建的四个和五个聚类混合物(图2a)。
如图显示了通过以不同比例混合两个转移样品而形成的计算机计算机混合物。底部图显示了创建四簇和五簇混合物的方法,该方法将每个混合物样本分成偶数和奇数染色体,然后对这些样本进行子采样以创建其他簇。最终的CCF基于每个奇数或偶数染色体样本的子采样百分比,而不是样本比例。
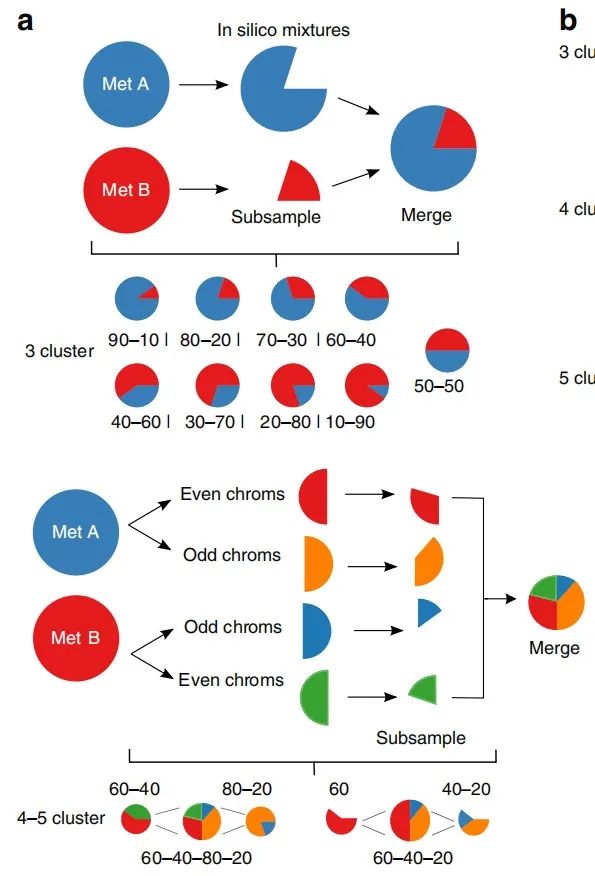
图2a. 用于创建真实肿瘤样品的计算机模拟混合物的二次采样和合并过程
四、最佳(Optimal)癌细胞比例与实际情况的对比
计算机模拟混合物使作者能够探索CCF分布的一些基本噪声特性(noise properties)。由于支持作者混合样本中的SV和SNV的读取计数易受噪声(近似二项分布)的影响,因此作者假设得出的CCF估计值也必须是嘈杂的(noisy,近似正态分布)。
为了观察到这一点,作者估计了每个变体的“最佳optimal” CCF,这是使用VAF计算的,并从真实(ground truth)的混合比例推断出多重性。该估计值使作者能够观察到最佳的CCF分布(图2c),在变异水平上,SV的CCF与SNV相比,平均误差(ME)略高。
ground truth:表示有监督学习的训练集的分类准确性,用于证明或者推翻某个假设。有监督的机器学习会对训练数据打标记,试想一下如果训练标记错误,那么将会对测试数据的预测产生影响,因此这里将那些正确打标记的数据成为ground truth。
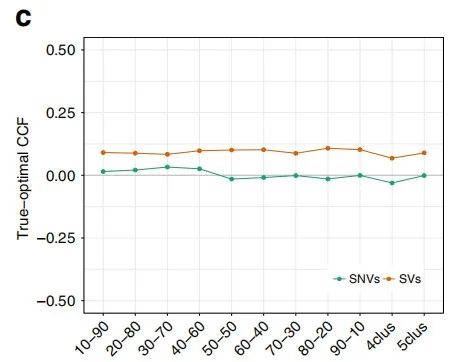
图2c. 最佳SNV和SV CCF与预期的ground truth CCF的平均每变量CCF误差
经过上述计算,得出的结果表明,作者观察到亚克隆聚类大致呈正态分布,而具有相似CCF的聚类具有重叠分布(图2b),较少的SV导致最佳的CCF估算值具有比SNV更少的清晰CCF峰,这些数据凸显了与SNV相比估算SV的CCF的难度。其中,ground truth CCF基于样本成员,最佳SV和SNV结果基于来自真实聚类均值的转换后的等位基因频率。
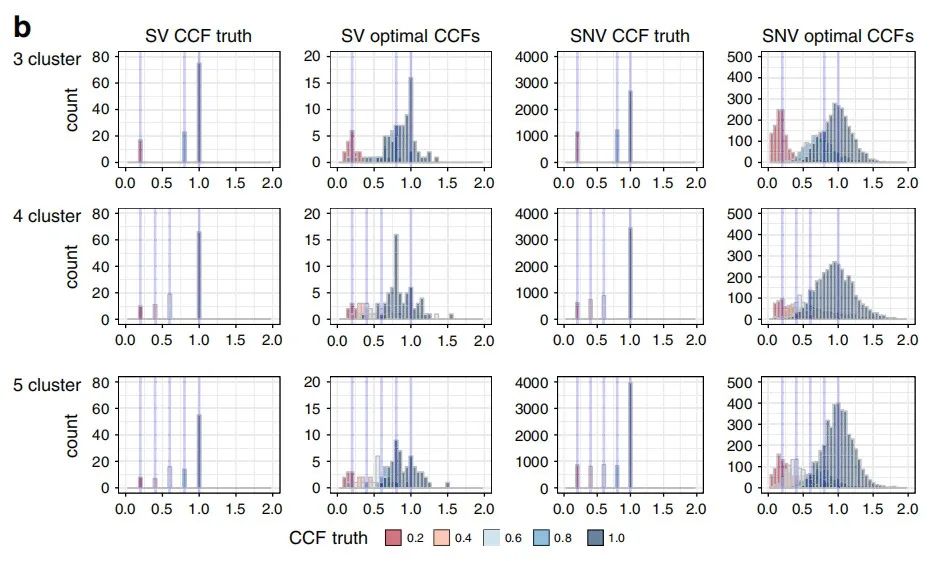
图2b. ground truth CCF与最佳SV和SNV结果相对于代表性的三、四和五聚类混合物
给定ground truth,这些数据还使作者能够确定用于确定变异是否为亚克隆的最佳每个变异临界值(cutoff)(图2d)。作者发现,采用两个SV末端的最大或平均CCF会得到最高的AUC(〜0.90),这也大约等于通过对SNV进行分类而获得的AUC。确定SV(使用平均CCF)和SNV的亚克隆性的最佳CCF截止值分别为0.69和0.72,为简化此,作者在所有下游分析中将两种变异类型的截止值均设为0.7。
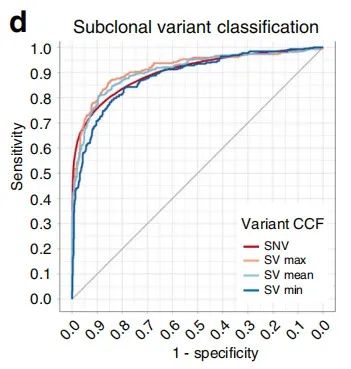
图2d. ROC曲线基于最佳变异CCF分为克隆或亚克隆
五、绩效评估(Performance assessment)
SVclone经过设计可确定单个全基因组测序肿瘤样品中SV的CCF。这些数据的常见下游分析包括分析样品中亚克隆种群的数量,和观察确认哪些SV是克隆或亚克隆的。因此,作者设计了性能指标来查询这些变量,包括:簇数错误,平均簇CCF错误,平均变体CCF错误以及调用变体亚克隆的敏感性和特异性。由于任何CCF推断算法的关键特征之一就是估计变异体的染色体拷贝数(称为多重性multiplicity),作者还观察到平均多重性误差。
目前没有用于估计SV CCF的方法可以直接比较,所以作者选择与两种代表性的最新方法进行比较,这些方法用于从单个样本中估算SNV的CCF:PyClone,拷贝数copy number,和Battenberg。此外,作者还以SNV集群模式运行SVclone,该模式使用Ccube的集群模型。性能总结在图3中,每种性能下的性能细分可以在图中找到。其中,SVclone的SV和SNV模型的性能,与在计算机混合物上运行的Battenberg(SCNR)和PyClone(SNV)相比。
-
第一列显示群集号错误(Cluster number error,三个推断的群集号)和平均CCF错误(Mean cluster CCF error),其中真实群集和推断的群集根据其顺序进行匹配。
-
群集号错误:此度量标准指示给定的簇算法在推断正确的簇数方面的有效性。应用于计算机混合物的SVclone能够在11例病例中的7例中识别出正确的簇数。与PyClone的4/11案例相比,SVclone的SNV集群在5/11案例中发现了正确数量的集群,这表明SV在识别正确数量的基础集群方面可能略有优势。
-
平均CCF错误:SV数据中的平均聚类CCF误差通常更高,平均平均误差为0.0913,而SVclone和PyClone分别在SNV数据中观察到0.0412和0.0756。这可能是由于变量数量差异造成的,因为SNV的数量相对较大,可能导致更准确的集群CCF估算
-
第二列显示了与基本真实CCF相比的平均变异CCF和多重误差。
-
平均变异CCF:SV数据中的平均变体CCF误差比其他方法略高,考虑到最优(即在已知聚类均值的情况下可获得的最佳结果),SV CCF的误码率会略高一些,分别为SV和SNV的均值分别为0.0408和0.002(图2c)。尽管就三簇混合物的平均变体CCF误差而言,Battenberg的平均表现要好于SVclone,但四簇和五簇混合物的SVclone表现更好,证明了SVclone能够考虑>2个亚克隆的优势。SVclone的SNV聚类和PyClone在混合物中显示出相似的平均误差趋势。
-
多重误差:多重误差表示从聚类推断出的多重性与给定真实CCF聚类均值的推断多重性之间的差异(因为不能直接观察到多重性)。SV中较低的多重错误率很可能归因于亚克隆拷贝数推断模型(仅考虑具有克隆拷贝数的SNV),该模型允许使用非整数拷贝数。三簇混合物中克隆SV的平均多重误差的绝对值近似于SNV多重误差。 由于PyClone在所有可能的多重性上取平均值,并且不直接估算多重性,因此作者没有将PyClone视为该指标。
-
第三列使用变异的样本成员资格显示亚克隆的分类敏感性和特异性(即,如果混合物的两个样品中均存在变异,则将其分类为克隆,否则为亚克隆)。
-
SVclone的SV估计值将变体分类为亚克隆时,对SNV的敏感性相似,平均敏感性为较高,SV的特异性较低;与其他方法相比,PyClone的灵敏度较低,但特异性更高;Battenberg具有最高的平均灵敏度和特异性,可以用Battenberg预期推断每个拷贝数分数的数据点(胚系SNV,germline SNVs)的数量。

图3. 集群性能指标与现有方法的比较
SVclone的性能可与基于SNV的聚类媲美,这表明尽管变异数量相对不足,但克隆结构仍可以高效,高一致性和准确性地进行重构。这意味着可以从SNV和SV分别推断出肿瘤的克隆结构,并比较其结果。
但是,如果假定样本中的克隆种群共享相同的SNV和SV,则作者还提供了使用相同的聚类框架对SV和SNV进行聚类的选项。当考虑基于模型的后期分配时,此功能更加强大。由补充资料可知,SNV CCF后验(posterior)可以与SV读取计数的可能性相结合,以进行分配调用,反之亦然。通过组合这些数据类型,可以提高整体性能。SVclone的两个独特设计功能还需要进一步的性能评估:
- SVclone将两个断点的后台SCNA状态合并到其群集模型中;
- SVclone在克隆和亚克隆拷贝数区域内聚集变体。
在这里,作者试图量化两种方法相对于“单纯”方法的优势,“单纯”方法只考虑每个SV一个断点,或者在克隆拷贝数区域仅用变异部分。
为了将SVclone的双端(dual-end)集群模型的性能与单个端(single end)进行比较,作者通过SVclone的单端(SNV)模型从三集群计算机模拟混合物中运行了相应的SV端,并将集群性能与双端模型比较。性能在图4中进行了汇总。
图4显示,在几乎所有混合(mixes)中——平均变异CCF误差,平均多重误差和均质CCF误差——双端模型均优于单端模型;与正确的单端模型相比,仅错误地推断出50–50混合的簇数。但是,考虑到50–50混合物的分裂,作者预计只有两个簇有效,因此双端模型的结果很可能与数据更省时。
- 有趣的是,单端模型显示出更高的亚克隆分类敏感性,但比双端模型具有更低的特异性。考虑到该指标代表了灵敏度和特异性的折衷,由补充材料得知,作者生成了ROC曲线,考虑到双端模型的AUC大于单端模型,作者表示最好使用双端模型。其中,SVclone的性能使用SV的两个端点或单个端点在三集群的计算机硅混合物上运行。第一列显示群集号错误(三个推断的群集号)和平均CCF错误,其中真实群集和推断的群集根据其顺序进行匹配。第二列显示了与基本真实CCF相比的平均变异CCF和多重误差。第三列使用变异的样本成员资格显示亚克隆的分类敏感性和特异性(即如果混合物的两个样品中均存在变异,则将其分类为克隆,否则为亚克隆)。
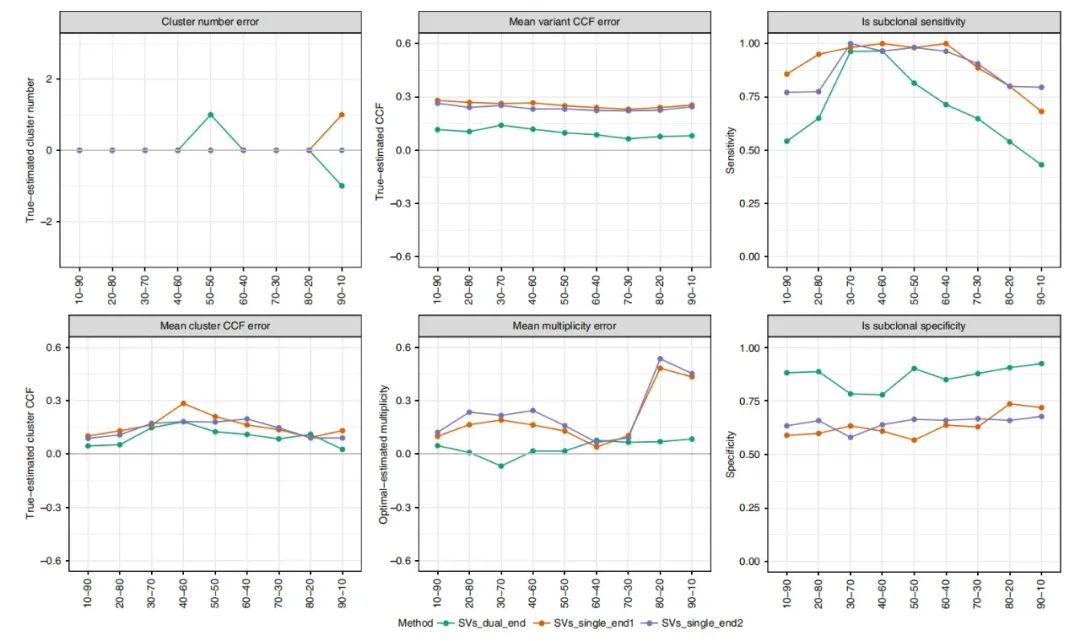
图4. 双端和单端模型的SV群集性能比较
最后,作者使用SV将SVclone的性能与克隆和亚克隆拷贝数区域进行了比较,图5中总结了性能。与仅具有克隆背景拷贝数状态的聚类SV相比,利用所有可用SV在所有指标上(除亚克隆分类特异性外)显着提高了性能。其中,SVclone的性能使用克隆背景拷贝数状态或克隆加亚克隆状态跨越三簇计算机模拟混合物。第一列显示群集号错误(三个推断的群集号)和平均CCF错误,其中真实群集和推断的群集根据其顺序进行匹配。第二列显示了与基本真实CCF相比的平均变异CCF和多重误差。第三列使用变异的样本成员资格显示亚克隆的分类敏感性和特异性(即如果混合物的两个样品中均存在变异,则将其分类为克隆,否则为亚克隆)。
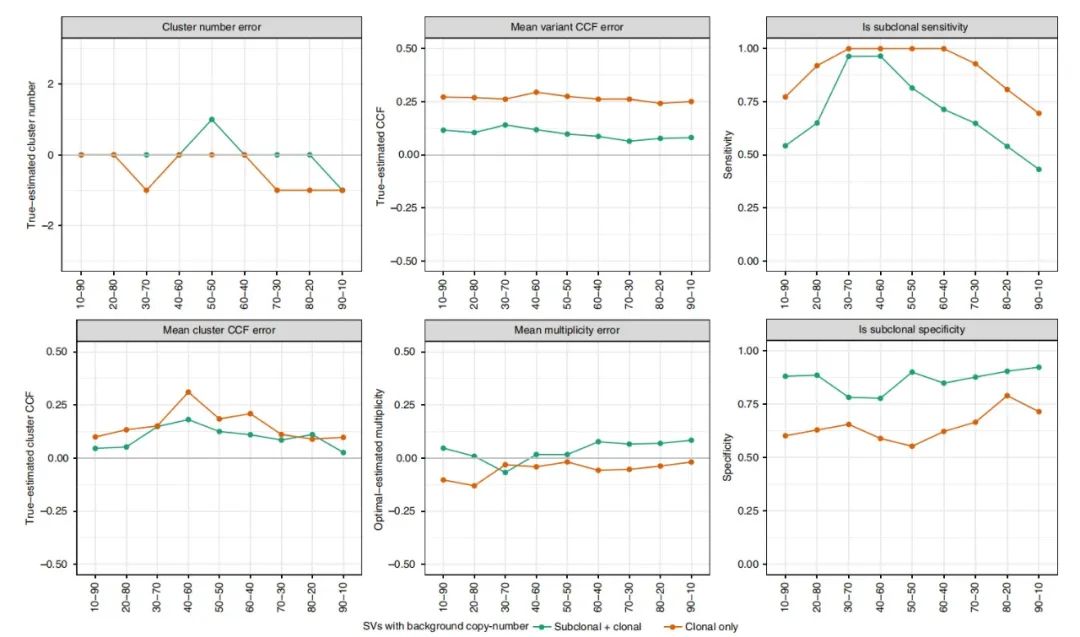
图5. SV聚类性能结合了背景亚克隆拷贝数状态
六 1705个全基因组测序肿瘤的克隆性分析
作者将SVclone应用于全基因组全癌癌分析(PCAWG)项目(dcc.icgc.org/ pcawg)的1705个WGS样品中,分别将SV和SNV进行聚类,试图观察SV与SNV相比克隆结构的任何差异。对23种肿瘤类型进行了下游分析,显示≥20个样本,SV> 10,SNV> 10,并且有足够的能力检测亚克隆性的总计n = 1169,亚克隆SV与SNV分数的比较显示了不同肿瘤类型的不同模式(图6a)。与SNV相比,亚克隆SV所占比例更高的肿瘤类型包括100%的肺鳞状细胞癌和92%的大肠腺瘤和卵巢腺癌。一些癌症还包含具有不同克隆模式的样本子集,例如,肝癌包含19个样本的群集,这些样本具有较高的SV亚克隆性(≥50%)和较低的SNV亚克隆性(<30%),其中,0.7 CCF下的变异被认为是亚克隆。
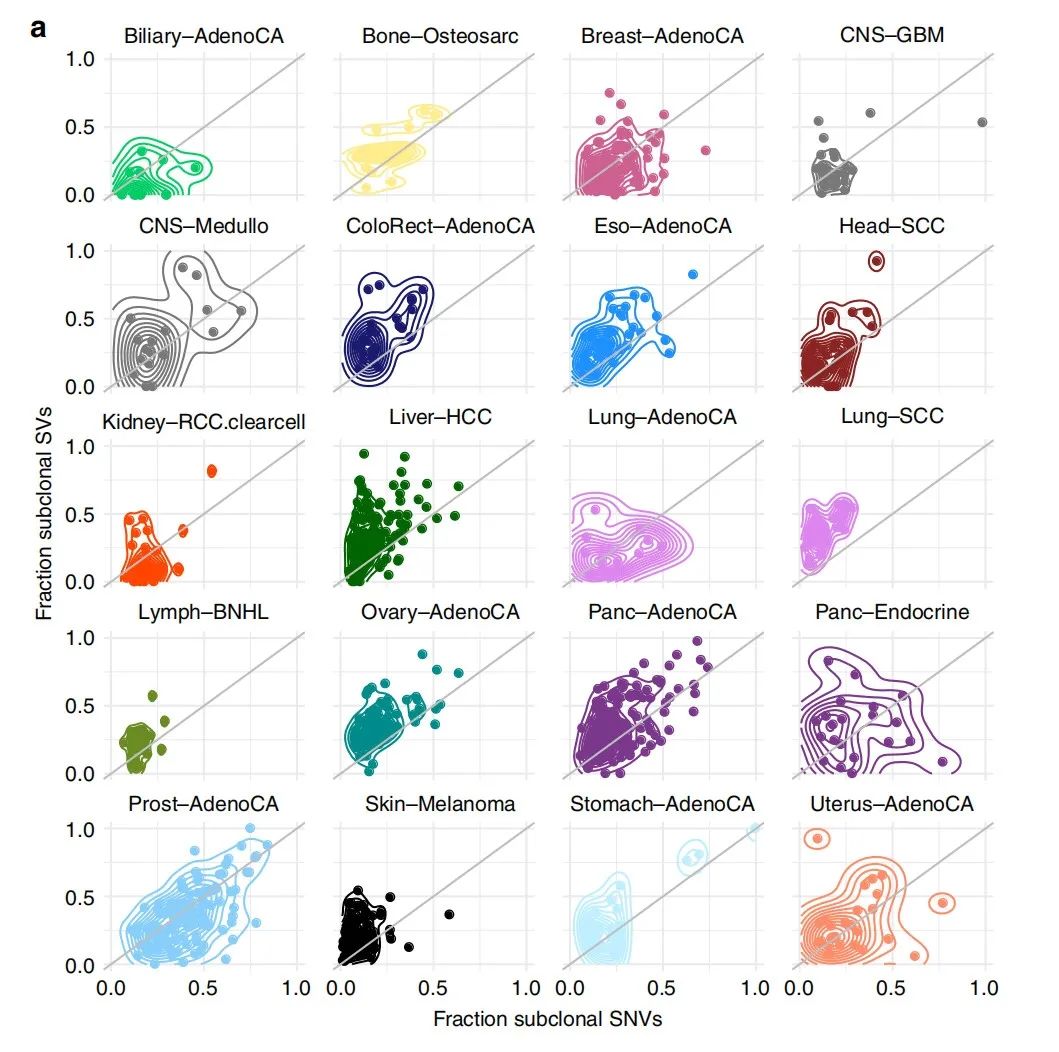
图6a. PCAWG样本(n = 1169)的亚克隆SV分数与SNV分数的二维密度图
SVclone的一个独特功能是,它确定拷贝数中性重排(反转和染色体间易位)的克隆性。作者对PCAWG队列中的亚克隆拷贝数中性重排进行了富集测试,共有28种癌症类型的177个样本显示了亚克隆拷贝数中性重排(SCNR)模式(图6c-f),其中卵巢癌(n = 29,占总卵巢样本的25.7%),肝细胞癌(n = 26,占肝样品的10.4%)和胰腺癌(n = 18,占胰腺样本的7.5%)。图6c中,外部轨道代表整个基因组的拷贝数,内部线代表SV。蓝线代表克隆SV,红线代表亚克隆SV。
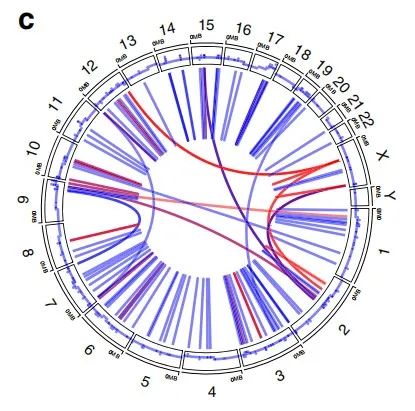
图6c. 示例SCNR模式肿瘤(肝细胞癌,肿瘤WGS等分试样)的圆图
其中,样本亚克隆SV的颜色按SV类别进行了编码。
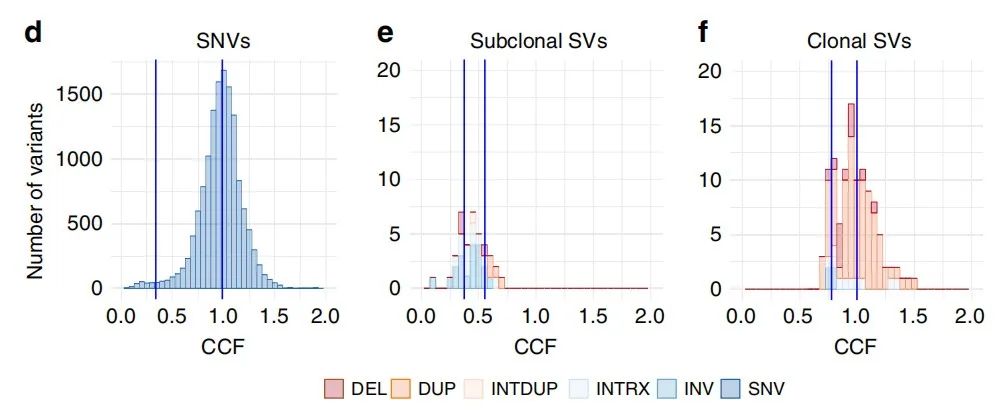
图6c-f. 样本的SNV,亚克隆,克隆SV的CCF直方图
为了测试这种SCNR模式的潜在临床相关性,作者比较了SCNR病例(n = 177),高SV异质性病例(n = 650)和所有其余病例(n =447),记录了总生存期,并根据年龄,肿瘤组织学亚型和SV数量进行了分层。这些组的生存概率显着不同,中位生存时间分别为1236、1470和2907天(图6b)。又由补充资料得知,作者没有发现富集折返倒置(FBI)的证据,这表明SCNR基因型可能来自于独立的机制。
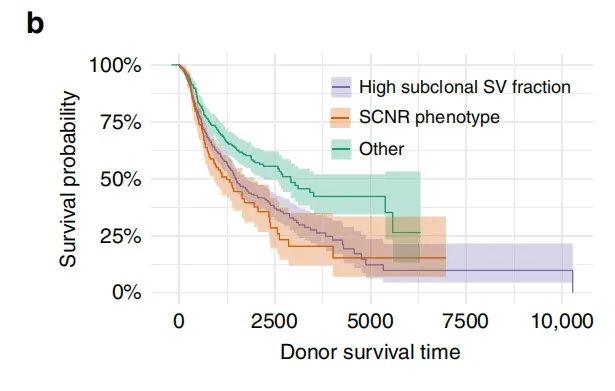
图6b. 患者的生存曲线分为具有SCNR模式的患者,具有高亚克隆SV分数的患者,或两者都不存在
为了测试这些SCNR事件是否是单个复杂的重排事件的结果,还是仅仅是一组不相关的重排,作者寻找聚类事件,并在可能的情况下尝试遍历衍生染色体。总体而言,这些聚集的SCNR事件中有50%与至少一个染色体间易位有关,而其他样本中只有22%,表明这些事件可以跨越多个染色体,加上双向t检验等补充实验,数据还表明SCNR样品中存在的亚克隆事件可能富含复杂,相互关联的重排。
为了提供有关SCNR基因型的病因学的一些见解,作者寻找了可能导致SCNR基因型的已知癌症驱动因素中SNV / INDEL的富集。具体来说,作者考虑了克隆(CCF> 0.7)突变,因为它们可能揭示了SCNR基因型的诱因。作者发现TP53突变(占SCNR样品的40.11%)与背景(占其他样品的14.54%)相比,含量更高。TP53的富集与所报道的TP53突变与复杂重排之间的联系相一致。然而,在高SV异质性队列(高SV异质性样本的36.77%)中,还观察到TP53 SNV / INDEL的富集,以及KRAS和CTNNB1,表明TP53可能是必需的,但不足以用于SCNR基因型。
最后,为了确定SCNR样品中亚克隆中性SV的富集是否包含功能序列,作者鉴定了所有具有SCNR候选双等位基因命中基因的驱动基因,并认定候选双等位基因(bi-allelic)hits是影响同一基因的两个单独的突变事件(拷贝数丢失,基因体内的SV和/或SNV / INDEL)。
七、小结
本文作者介绍了一种推断结构变异断点的癌细胞分数的综合方法SVclone,并证明了考虑中性重排的克隆性的重要性,本文的关键在于,在这项工作中,聚类模型将所有SV视为独立事件,尽管在某些情况下这些SV可能是同一复杂重排的一部分。SVclone的局限性在于其分类框架无法识别复杂的重排,但是用户可以自己指定类型。
参考资料
- https://cloud.tencent.com/developer/article/1677644
- Inferring structural variant cancer cell fraction
