【2.6.2】VarScan肿瘤体细胞突变检测
VarScan是一个java写的linux系统下进行肿瘤体细胞突变检测(calls SNV in somatic variants)的软件,可用于目标捕获(targeted),外显子( exome)以及全基因组测序(whole-genome resequencing) 等多种测序数据。
一、简介
插一句:体细胞突变(somatic mutation)是指患者某些组织或者器官后天性地发生了体细胞变异,虽然它不会遗传给后代个体,却可以通过细胞分裂,遗传给子代细胞。体细胞突变对肿瘤的发生发展有关键性的作用,并且它也是制定肿瘤癌症靶向治疗措施的关键所在。
VARSCAN有3个功能,如下图:
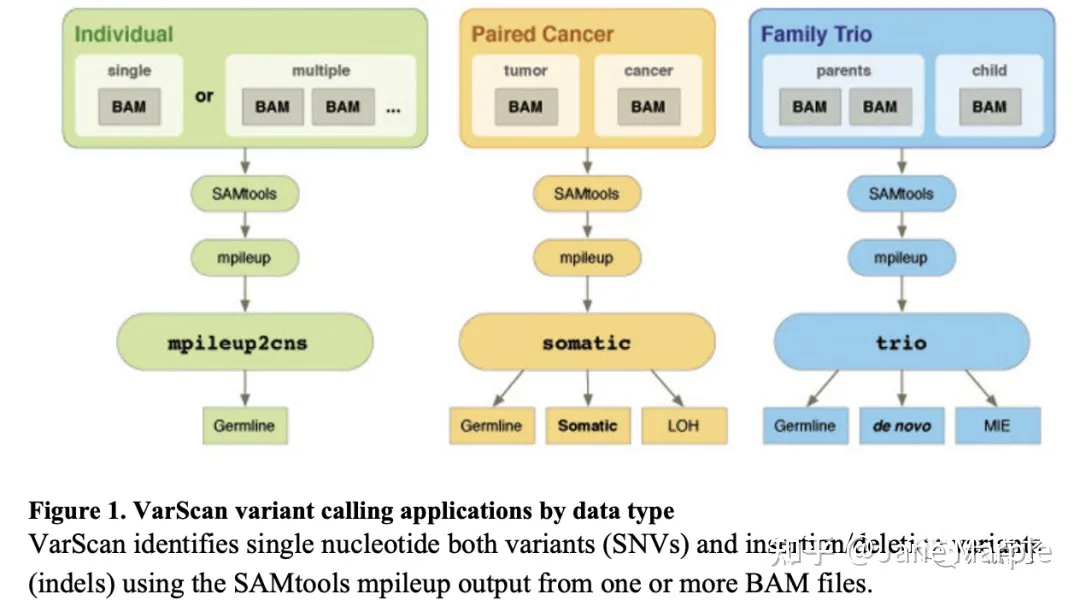
比较常用的是肿瘤样本——正常样本配对somatic mutation模式。
二、安装说明
2.1. 安装
VarScan寄存在sourceforge网站[1],可通过链接:https://sourceforge.net/projects/varscan/files/
下载java二进制文件:
wget --no-check-certificate https://nchc.dl.sourceforge.net/project/varscan/VarScan.v2.3.9.jar
这里我们指定–no-check-certificate 允许chc.dl.sourceforge.net的过期证书。
终端运行:
# java -jar VarScan.jar
VarScan v2.3
USAGE: java -jar VarScan.jar [COMMAND] [OPTIONS]
COMMANDS:
pileup2snp Identify SNPs from a pileup file
pileup2indel Identify indels a pileup file
pileup2cns Call consensus and variants from a pileup file
mpileup2snp Identify SNPs from an mpileup file
mpileup2indel Identify indels an mpileup file
mpileup2cns Call consensus and variants from an mpileup file
somatic Call germline/somatic variants from tumor-normal pileups
copynumber Determine relative tumor copy number from tumor-normal pileups
readcounts Obtain read counts for a list of variants from a pileup file
filter Filter SNPs by coverage, frequency, p-value, etc.
somaticFilter Filter somatic variants for clusters/indels
fpfilter Apply the false-positive filter
processSomatic Isolate Germline/LOH/Somatic calls from output
copyCaller GC-adjust and process copy number changes from VarScan copynumber output
compare Compare two lists of positions/variants
limit Restrict pileup/snps/indels to ROI positions
安装成功。
2.2 软件运行前准备工作-pileup文件
VarScan需要准备正常样本,肿瘤样本pileup文件, 首先,使用samtools处理normal,tumor样本的bam文件,得到相应pileup文件。
运行命令:
samtools mpileup \
-q 1 \
-f $REFERENCE \
${NORMAL_BAM} \
--output $WORKDIR/tmp/$SAMPLE.$prefix.normal.pileup
samtools mpileup \
-q 1 \
-f $REFERENCE \
${TUMOR_BAM} \
--output $WORKDIR/tmp/$SAMPLE.$prefix.tumor.pileup
2.3 软件运行
运行
java -Xmx10g -jar /home/6oclock/bin/VarScan.v2.3.9.jar somatic \
$WORKDIR/tmp/$SAMPLE.$prefix.normal.pileup \
$WORKDIR/tmp/$SAMPLE.$prefix.tumor.pileup \
$WORKDIR/tmp/$SAMPLE.$prefix.mut
输出文件:运行完会生成.snp和.indel文件,其中后缀为.snp的文件即为对应snv结果,.indel为indel结果。
三、实战
以NA12878样本的测序数据,具体来实践一下吧。
3.1 数据下载
下载肿瘤样本与配对的正常样本的测序bam数据:
# 网站站点
data_url=http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/WGS
# 正常样本
wget ${data_url}/NA12878.WGS.chrM.BwaMem.sort.bam
# 肿瘤样本
wget ${data_url}/hg19.NA12878.tumor_addsnv.WGS.chrM.BwaMem.bam
下载比对需要的参考基因组数据及其索引文件:
# 网站站点
data_url=http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/Reference/hg19.chrM.fasta_BwaMem_index
# 参考基因
wget ${data_url}/hg19.chrM.fasta
# 索引
wget ${data_url}/hg19.chrM.dict
wget ${data_url}/hg19.chrM.fasta.amb
wget ${data_url}/hg19.chrM.fasta.ann
wget ${data_url}/hg19.chrM.fasta.bwt
wget ${data_url}/hg19.chrM.fasta.pac
wget ${data_url}/hg19.chrM.fasta.sa
3.2 准备pileup文件
使用samtools分别处理normal,tumor样本的bam文件,以生成相应pileup文件, shell运行:
# 处理正常样本
samtools mpileup \
-q 1 \
-f hg19.chrM.fasta \
NA12878.WGS.chrM.BwaMem.sort.bam \
--output NA12878.normal.pileup
# 处理肿瘤样本
samtools mpileup \
-q 1 \
-f hg19.chrM.fasta \
hg19.NA12878.tumor_addsnv.WGS.chrM.BwaMem.bam \
--output NA12878.tumor.pileup
3.3 运行命令
此处我们将程序运行的标准输出和标准错误都分别重定向到对应的log和err文件中了。一步式运行:
java -Xmx10g -jar VarScan.v2.3.9.jar somatic \
NA12878.normal.pileup \
NA12878.tumor.pileup \
NA12878.tumor \
1>ass.log \
2>ass.err
3.4 输出结果
此处我们的测试案例数据做了截取,因此可以非常快速的跑完,具体的结果如下图所示,可以看到生成了NA12878.tumor.snp 和NA12878.tumor.indel文件。其中NA12878.tumor.snp 即为最终检测出的肿瘤SNV结果,结构如下所示:
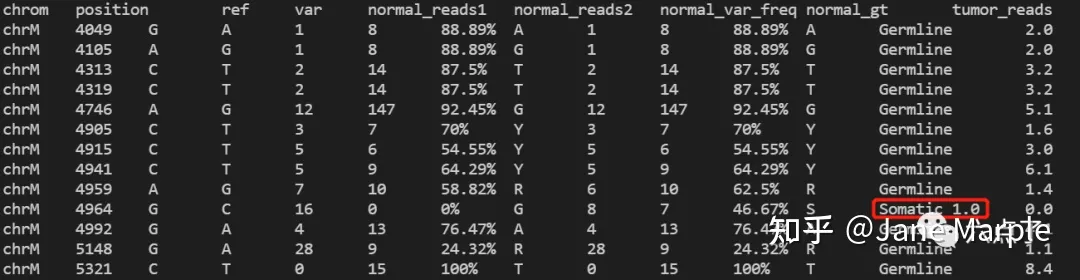
输出结果中几个比较重要的列的解释:
- chrom :染色体;
- position :染色体坐标;
- ref :参考基因该位点基因型。
- var :变异位点的基因型;
- normal_gt:正常样本该位点基因型;
- tumor_gt:肿瘤样本该位点基因型;
- somatic_status : 该变异的类型,一般区分为Germline和Somatic ,如上图标红部分,即为一个体细胞突变(somatic mutation) 。
至此,VarScan的实战体验基本就结束了。
四、sixbox运行
此外,sixoclock官网[2]基于CWL (common workflow language) 对VarScan软件进行了封装,通过我们开发的sixbox 软件可以快速进行软件的运行。对sixbox不了解可以通过六点了官网[3]了解下。下面是具体的运行步骤如下。
下载cwl 源码
sixbox pull c6e4a52a-ecb4-470d-90c1-0d2fcbd42e09
运行sixbox cwls 可以看到下载成功:
下载数据
wget http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/WGS/NA12878_addsnv.tumor.pileup
wget http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/WGS/NA12878_addsnv.normal.pileup
参数配置
使用sixbox生成参数模板文件(YAML) , 并配置yaml文件
sixbox run --make-template 6oclock/VarScan-Somatic:v2.3.9 > varscan.job.yaml
vim varscan.job.yaml # 编辑参数配置文件,替换或设置参数以实现个性化分析
可以直接粘贴下方示例内容到varscan.job.yaml
out_file_name: NA12878_addsnv
tumor_pileup: # type "File"
class: File
path: http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/WGS/NA12878_addsnv.tumor.pileup
normal_pileup: # type "File"
class: File
path: http://www.sixoclock.net/resources/data/NGS/Homo_sapiens/WGS/NA12878_addsnv.normal.pileup
使用sixbox运行 运行命令,
sixbox run 6oclock/VarScan-Somatic:v2.3.9 varscan.job.yaml
即可看到当前目录或者指定的输出目录输出对应的结果文件。运行日志如下:
{
"snp_file": {
"location": "file:///home/6oclock/project/cwlTests/vanscan_somatic_out.snp",
"basename": "vanscan_somatic_out.snp",
"class": "File",
"checksum": "sha1$d4866a3b95da941fcd10f3543075996c75b00a6b",
"size": 5620,
"path": "/home/6oclock/project/cwlTests/vanscan_somatic_out.snp"
},
"indel_file": {
"location": "file:///home/6oclock/project/cwlTests/vanscan_somatic_out.indel",
"basename": "vanscan_somatic_out.indel",
"class": "File",
"checksum": "sha1$15e45c3b0a3287b82ad7278e09a2f573c85784e3",
"size": 326,
"path": "/home/6oclock/project/cwlTests/vanscan_somatic_out.indel"
}
}
INFO Final process status is success
以上为我们给大家带来的肿瘤基因组体细胞的基本原理知识,以及在六点了平台上运行经典的VanScan的详细操作过程。如果对生物医疗健康大数据相关内容感兴趣也可以持续关注我们。
参考资料
- https://zhuanlan.zhihu.com/p/473757553
- sourceforge网站: http://varscan.sourceforge.net/
- sixoclock官网: http://www.sixoclock.net
- 六点了官网: http://www.sixoclock.net
- Koboldt, D., Zhang, Q., Larson, D., Shen, D., McLellan, M., Lin, L., Miller, C., Mardis, E., Ding, L., & Wilson, R. (2012). VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing Genome Research DOI: 10.1101/gr.129684.111
