【4.1.4】基因突变的功能注释--VEP
VEP(Variant Effect Predictor)官网教程:
- https://grch37.ensembl.org/info/docs/tools/vep/script/vep_tutorial.html
- https://www.ensembl.org/info/docs/tools/vep/script/vep_options.html
一、软件简介
VEP(Variant Effect Predictor):ensembl旗下的注释工具,属于比较常用的注释软件
https://asia.ensembl.org/info/docs/tools/vep/index.html
基本功能:
1)借助Sequence Ontology (SO)将突变类型大致分为39种类型 https://asia.ensembl.org/info/genome/variation/prediction/predicted_data.html#consequences

| * | SO term | SO description | SO accession | Display term | IMPACT |
|---|---|---|---|---|---|
| transcript_ablation | A feature ablation whereby the deleted region includes a transcript feature | SO:0001893 | Transcript ablation | HIGH | |
| splice_acceptor_variant | A splice variant that changes the 2 base region at the 3' end of an intron | SO:0001574 | Splice acceptor variant | HIGH | |
| splice_donor_variant | A splice variant that changes the 2 base region at the 5' end of an intron | SO:0001575 | Splice donor variant | HIGH | |
| stop_gained | A sequence variant whereby at least one base of a codon is changed, resulting in a premature stop codon, leading to a shortened transcript | SO:0001587 | Stop gained | HIGH | |
| frameshift_variant | A sequence variant which causes a disruption of the translational reading frame, because the number of nucleotides inserted or deleted is not a multiple of three | SO:0001589 | Frameshift variant | HIGH | |
| stop_lost | A sequence variant where at least one base of the terminator codon (stop) is changed, resulting in an elongated transcript | SO:0001578 | Stop lost | HIGH | |
| start_lost | A codon variant that changes at least one base of the canonical start codon | SO:0002012 | Start lost | HIGH | |
| transcript_amplification | A feature amplification of a region containing a transcript | SO:0001889 | Transcript amplification | HIGH | |
| feature_elongation | A sequence variant that causes the extension of a genomic feature, with regard to the reference sequence | SO:0001907 | Feature elongation | HIGH | |
| feature_truncation | A sequence variant that causes the reduction of a genomic feature, with regard to the reference sequence | SO:0001906 | Feature truncation | HIGH | |
| inframe_insertion | An inframe non synonymous variant that inserts bases into in the coding sequence | SO:0001821 | Inframe insertion | MODERATE | |
| inframe_deletion | An inframe non synonymous variant that deletes bases from the coding sequence | SO:0001822 | Inframe deletion | MODERATE | |
| missense_variant | A sequence variant, that changes one or more bases, resulting in a different amino acid sequence but where the length is preserved | SO:0001583 | Missense variant | MODERATE | |
| protein_altering_variant | A sequence_variant which is predicted to change the protein encoded in the coding sequence | SO:0001818 | Protein altering variant | MODERATE | |
| splice_donor_5th_base_variant | A sequence variant that causes a change at the 5th base pair after the start of the intron in the orientation of the transcript | SO:0001787 | Splice donor 5th base variant | LOW | |
| splice_region_variant | A sequence variant in which a change has occurred within the region of the splice site, either within 1-3 bases of the exon or 3-8 bases of the intron | SO:0001630 | Splice region variant | LOW | |
| splice_donor_region_variant | A sequence variant that falls in the region between the 3rd and 6th base after splice junction (5' end of intron) | SO:0002170 | Splice donor region variant | LOW | |
| splice_polypyrimidine_tract_variant | A sequence variant that falls in the polypyrimidine tract at 3' end of intron between 17 and 3 bases from the end (acceptor -3 to acceptor -17) | SO:0002169 | Splice polypyrimidine tract variant | LOW | |
| incomplete_terminal_codon_variant | A sequence variant where at least one base of the final codon of an incompletely annotated transcript is changed | SO:0001626 | Incomplete terminal codon variant | LOW | |
| start_retained_variant | A sequence variant where at least one base in the start codon is changed, but the start remains | SO:0002019 | Start retained variant | LOW | |
| stop_retained_variant | A sequence variant where at least one base in the terminator codon is changed, but the terminator remains | SO:0001567 | Stop retained variant | LOW | |
| synonymous_variant | A sequence variant where there is no resulting change to the encoded amino acid | SO:0001819 | Synonymous variant | LOW | |
| coding_sequence_variant | A sequence variant that changes the coding sequence | SO:0001580 | Coding sequence variant | MODIFIER | |
| mature_miRNA_variant | A transcript variant located with the sequence of the mature miRNA | SO:0001620 | Mature miRNA variant | MODIFIER | |
| 5_prime_UTR_variant | A UTR variant of the 5' UTR | SO:0001623 | 5 prime UTR variant | MODIFIER | |
| 3_prime_UTR_variant | A UTR variant of the 3' UTR | SO:0001624 | 3 prime UTR variant | MODIFIER | |
| non_coding_transcript_exon_variant | A sequence variant that changes non-coding exon sequence in a non-coding transcript | SO:0001792 | Non coding transcript exon variant | MODIFIER | |
| intron_variant | A transcript variant occurring within an intron | SO:0001627 | Intron variant | MODIFIER | |
| NMD_transcript_variant | A variant in a transcript that is the target of NMD | SO:0001621 | NMD transcript variant | MODIFIER | |
| non_coding_transcript_variant | A transcript variant of a non coding RNA gene | SO:0001619 | Non coding transcript variant | MODIFIER | |
| coding_transcript_variant | A transcript variant of a protein coding gene | SO:0001968 | Coding transcript variant | MODIFIER | |
| upstream_gene_variant | A sequence variant located 5' of a gene | SO:0001631 | Upstream gene variant | MODIFIER | |
| downstream_gene_variant | A sequence variant located 3' of a gene | SO:0001632 | Downstream gene variant | MODIFIER | |
| TFBS_ablation | A feature ablation whereby the deleted region includes a transcription factor binding site | SO:0001895 | TFBS ablation | MODIFIER | |
| TFBS_amplification | A feature amplification of a region containing a transcription factor binding site | SO:0001892 | TFBS amplification | MODIFIER | |
| TF_binding_site_variant | A sequence variant located within a transcription factor binding site | SO:0001782 | TF binding site variant | MODIFIER | |
| regulatory_region_ablation | A feature ablation whereby the deleted region includes a regulatory region | SO:0001894 | Regulatory region ablation | MODIFIER | |
| regulatory_region_amplification | A feature amplification of a region containing a regulatory region | SO:0001891 | Regulatory region amplification | MODIFIER | |
| regulatory_region_variant | A sequence variant located within a regulatory region | SO:0001566 | Regulatory region variant | MODIFIER | |
| intergenic_variant | A sequence variant located in the intergenic region, between genes | SO:0001628 | Intergenic variant | MODIFIER | |
| sequence_variant | A sequence_variant is a non exact copy of a sequence_feature or genome exhibiting one or more sequence_alteration | SO:0001060 | Sequence variant | MODIFIER |
二、安装
方法1 通过conda来安装(我是通过这个方法安装好的):
实际上,使用conda安装vep是最快的 (最新的版本号是107, 个人需要所以安装100版本)
conda create -n vep
conda activate vep #我的是conda activate pVACtools
conda install -c bioconda ensembl-vep
#conda install -c bioconda ensembl-vep=100
注意:conda安装过程只安装了依赖的包和程序代码,数据库还需自己下载
方法2
官网教程推荐用git安装 (这个因为网络问题,以及库的冲突,搞了半天)
git clone https://github.com/Ensembl/ensembl-vep
cd ensembl-vep
git pull
git checkout release/107 #可以改成你需要的版本号
perl INSTALL.pl
方法3
## 2.1 首先安装cpanm:http://www.cpan.org/modules/INSTALL.html
cpan App::cpanminus
## Now install any module you can find.
## cpanm Module::Name
## 我的经验是:不要在conda环境中安装,就在普通环境中安装
cpanm Archive::Zip
cpanm DBD::mysql
cpanm DBI
cpanm Bio::SeqFeature::Lite
cpanm Module::Build
cpanm Try::Tiny
## 2.2 下载VEP
## 假设你的安装路径为:/home/user_name/software/ensembl-vep-release-107
curl -L -O https://github.com/Ensembl/ensembl-vep/archive/release/107.zip
unzip 107.zip
cd ensembl-vep-release-107/
## 2.3 新建目录和环境变量
mkdir /home/user_name/software/ensembl-vep-release-107/cache
mkdir /home/user_name/software/ensembl-vep-release-107/plugins
## 1. add /home/user_name/software/ensembl-vep-release-107 to your PERL5LIB environment variable
## 2. add /home/user_name/software/ensembl-vep-release-107/htslib to your PATH environment variable
export PATH=$PATH:/home/user_name/software/ensembl-vep-release-107/htslib
export PERL5LIB=$PERL5LIB:/home/user_name/software/ensembl-vep-release-10
## 2.4 安装VEP
## 必加参数:--DESTDIR
## 运行这个cmd:
## a (API + Bio::DB::HTS/htslib)
perl INSTALL.pl --AUTO a --CACHEDIR /home/user_name/software/ensembl-vep-release-107/cache --DESTDIR /home/user_name/software/ensembl-vep-release-107 --SPECIES homo_sapiens --ASSEMBLY GRCh37
## ==== 最后显示:
All tests successful.
Files=42, Tests=1768, 127 wallclock secs ( 0.43 usr 0.14 sys + 118.73 cusr 7.60 csys = 126.90 CPU)
Result: PASS
- OK!
## 然后运行这个cmd:
## c (cache)
perl INSTALL.pl --AUTO c --CACHEDIR /home/user_name/software/ensembl-vep-release-107/cache --DESTDIR /home/user_name/software/ensembl-vep-release-107 --SPECIES homo_sapiens --ASSEMBLY GRCh37
perl INSTALL.pl --AUTO c --CACHEDIR /home/user_name/software/ensembl-vep-release-107/cache --DESTDIR /home/user_name/software/ensembl-vep-release-107 --SPECIES homo_sapiens --ASSEMBLY GRCh38
## 然后运行这个cmd:
## p (plugins) — Require the use of the --PLUGINS flag to list the plugin(s) to install.
perl INSTALL.pl --ASSEMBLY GRCh37 --PLUGINS AncestralAllele,CADD,ClinPred,Conservation,dbNSFP,LoFtool --PLUGINSDIR /home/user_name/software/ensembl-vep-release-107/plugins --CACHEDIR /home/user_name/software/ensembl-vep-release-107/cache --SPECIES homo_sapiens
下载注释数据库
https://asia.ensembl.org/info/docs/tools/vep/script/vep_cache.html 建议cache 要跟vep版本匹配
VEP can use a variety of annotation sources to retrieve the transcript models used to predict consequence types.
- Cache - a downloadable file containing all transcript models, regulatory features and variant data for a species
- GFF or GTF - use transcript models defined in a tabix-indexed GFF or GTF file
- Database - connect to a MySQL database server hosting Ensembl databases
推荐使用相同版本的Cache: Cache: a downloadable file containing all transcript models, regulatory features and variant data for a species
我的命令:
cd /data2/neoantigen/annotation_data_grch38_ens95/vep_cache2
wget -c https://ftp.ensembl.org/pub/release-105/variation/indexed_vep_cache/homo_sapiens_vep_105_GRCh38.tar.gz
tar xzf homo_sapiens_vep_105_GRCh38.tar.gz
wget -c https://ftp.ensembl.org/pub/release-105/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
gzip -d Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
bgzip Homo_sapiens.GRCh38.dna.primary_assembly.fa
./vep -i input.vcf –offline –hgvs –fasta Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
其他方法:
方法一:使用vep命令
vep_install -a cf -s homo_sapiens -y GRCh37 -c /your_vepDB_dir/
# /usr/bin/perl /home/sam/project/neoantigen/lib/ensembl-vep/INSTALL.pl --CACHEDIR $BASEDIR/vep_cache --AUTO cf --SPECIES homo_sapiens --ASSEMBLY GRCh38
方法二:使用wget从vep的FTP下载
ftp://ftp.ensembl.org/pub/release-100/variation/indexed_vep_cache/homo_sapiens_vep_100_GRCh37.tar.gz
解压下载的tar.gz文件
tar -zxvf homo_sapiens_vep_100_GRCh37.tar.gz
测试:运行vep
./vep -i examples/homo_sapiens_GRCh38.vcf --cache
三、参数说明
vep -i homo_sapiens_GRCh38.vcf --fork 4 -o homo_sapiens_GRCh38.out.vcf \
--assembly GRCh38 --cache --cache_version 105 \
--dir /path/envs/vep/share/ensembl-vep-101.0-1(path) --offline \
--fasta /path/envs/vep/share/ensembl-vep-101.0-1/homo_sapiens/101_GRCh37/Homo_sapiens.GRCh37.dna.primary_assembly.fa.gz \
--force_overwrite
必需参数
--input_file / -i 输入文件
--output_file / -o 指定输出文件名称(默认为"variant_effect_output.txt" )
--cache 使用cache数据库进行注释
Cache相关参数
--dir_cache 下载的cache文件所在目录
--cache_version 指定cache版本
--offline 使用本地运行模式
在用参数
--fasta 指明所用的参考基因组所在位置(第一次使用这个参数时,会自动给基因组fasta文件创建index,时间较长)注:使用--hgvs选项时,必须使用--fasta
--everything 输出所有可用的注释条目,启用的参数包括(--sift b, --polyphen b, --ccds, --hgvs, --symbol, --numbers, --domains, --regulatory, --canonical, --protein, --biotype, --af, --af_1kg, --af_esp, --af_gnomade, --af_gnomadg, --max_af, --pubmed, --uniprot, --mane, --tsl, --appris, --variant_class, --gene_phenotype, --mirna)
--sift 用算法预测突变引起的氨基酸变化对蛋白的功能是否有害。预测结果分为"deleterious" or "tolerated"两种。 --sift b表示同时给出预测结果和分值。如果加上-filter "SIFT is deleterious" 则可以筛选出有害的(deleterious)变异
--polyphen 启用PolyPhen工具对氨基酸变异对蛋白结构和功能的影响进行注释。--polyphen b表示同时给出预测结果和分值
--ccds 输出结果加入CCDS列,显示对应的CCDS transcript
--hgvs 输出结果加入HGVS注释(必须与--fasta同时使用)
--symbol 输出结果加入SYMBOL列,显示基因名称
--numbers 加入突变所在外显子/内含子位置(第几号)
--domains DOMAINS列, Adds names of overlapping protein domains to output
--regulatory Look for overlaps with regulatory regions
--canonical 输出结果加入CANONICAL列,这个tag注明所使用的是否是canonical转录本
--protein 输出结果加入ENSP列,显示ENSP编号
--biotype BIOTYPE列,Adds the biotype of the transcript or regulatory feature
--af Add the global allele frequency (AF) from 1000 Genomes Phase 3 data for any known co-located variant to the output
--af_1kg 在注释中加入1000Genome的不同人群频率(必须与--cache同时使用)
--af_esp Include allele frequency from NHLBI-ESP populations(必须与--cache同时使用)
--af_gnomadg Include allele frequency from Genome Aggregation Database (gnomAD) genome populations(必须与--cache同时使用)
-max_af 报告出1000Genome数据库中最高的人群频率
--pubmed Report Pubmed IDs for publications that cite existing variant.
--uniprot 输出UniProt数据库中最匹配的一条记录的accessions
--mane 增加一个tag注明所用的transcript是否在 MANE Select
或MANE Plus Clinical数据库中(只适用于GRCh38基因组)
--tsl Adds the transcript support level for this transcript to the output(只适用于GRCh38基因组)
--appris Adds the APPRIS isoform annotation for this transcript to the output(只适用于GRCh38基因组)
--variant_class Output the Sequence Ontology variant class
--gene_phenotype Indicates if the overlapped gene is associated with a phenotype, disease or trait
--mirna Reports where the variant lies in the miRNA secondary structure
其他参数
--refseq 使用refseq数据库进行注释
--species 说明测序物种(如果不是human,则需下载对应物种的注释数据库)
--vcf 按照VCF标准格式进行输出
--no_stats 不产生统计信息
--fork 使用多个CPU并行计算
--force_overwrite 如果输出文件已经存在,则强制覆盖(简写为--force)
--config 可以把常用的配置写在文件中通过此参数调用
--fields 自定义输出的条目(多个条目间使用 , 分隔)
四、我得案例
4.1 常用命令
/usr/bin/perl /home/sam/project/neoantigen/lib/ensembl-vep/INSTALL.pl --CACHEDIR $BASEDIR/vep_cache --AUTO cf --SPECIES homo_sapiens --ASSEMBLY GRCh38
4.2 使用心得
建议用这个来确认:
或者选择ensembl
注释结果的进一步确认,尤其是 stop_gained 等状态的进一步确认
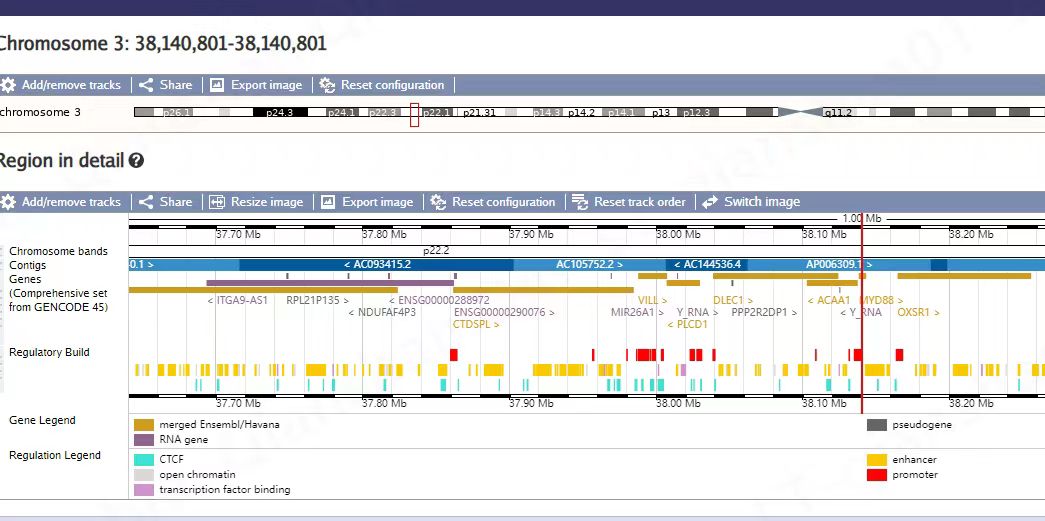
https://www.ensembl.org/Homo_sapiens/Location/View?r=3%3A38140803-38140803
https://asia.ensembl.org/Homo_sapiens/Location/View?r=3%3A38140803-38140803
这里有一个很奇怪的现象,我们用 vep_106_hg38 注释的话,这个点注释为ACAA1的上游基因。 漏掉了MYD88 这个位置,导致下游的分析受阻。最终通过ensembl上确认,才看明白怎么回事。
五、我的报错
报错1:
Can’t locate Module/Build.pm in @INC (you may need to install the Module::Build module) (@INC contains: /usr/local/lib/perl5/x86_64-linux-thread-multi /usr/local/lib/perl5 /usr/local/lib/perl5/x86_64-linux-thread-multi /usr/
解决办法:
cpan App::cpanminus
cpan Module::Build
perl -MCPAN -e 'install Bio::Perl'
cpanm autodie
cpanm Archive::Zip
cpanm DBD::mysql
cpanm DBI
cpanm Bio::SeqFeature::Lite # 失败
cpanm Module::Build
cpanm Try::Tiny
报错2
cpanm DBD::mysql 报错: Can‘t exec “mysql_config“: No such file or directory at Makefile.PL
http://www.cpan.org/authors/id/D/DV/DVEEDEN/DBD-mysql-4.050.tar.gz
解决办法:
yum install mysql # 因为没有装mysql, 所以这个库一直有问题
yum install mariadb-devel # 因为没有装mysql, 所以这个库一直有问题
cpanm DBD::mysql
手动安装 DBD::mysql
cd /data/src/
wget -c https://www.cpan.org/modules/by-module/DBD/DBD-mysql-4.032.tar.gz
gzip -cd DBD-mysql-4.032.tar.gz | tar xf -
cd DBD-mysql-4.032
perl Makefile.PL
make
make test
(On Windows you may need to replace ``make'' with ``nmake'' or ``dmake''.) If the tests seem to look fine, you may continue with
make install
https://perl.mines-albi.fr/perl5.6.1/site_perl/5.6.1/sun4-solaris/DBD/mysql/INSTALL.html
手动安装DBI
ERROR: DBI module not found. VEP requires the DBI perl module to function
#下载地址: http://www.cpan.org/modules/by-module/DBD/
wget -c http://www.cpan.org/modules/by-module/DBD/DBI-1.630.tar.gz
tar zxvf DBI-1.630.tar.gz
cd DBI-1.630
perl Makefile.PL
make
make test
make install
原文链接:https://blog.csdn.net/meegle/article/details/26093969
报错3:
./Bio/tmp/Bio-DB-HTS-2.11 - moving files to ./biodbhts
- making Bio::DB:HTS
/data/software/anaconda3/bin/x86_64-conda_cos6-linux-gnu-ld: unrecognized option '-Wl,-O2'
/data/software/anaconda3/bin/x86_64-conda_cos6-linux-gnu-ld: use the --help option for usage information
Warning: ExtUtils::CBuilder not installed or no compiler detected
Proceeding with configuration, but compilation may fail during Build
Created MYMETA.yml and MYMETA.json
Creating new 'Build' script for 'Bio-DB-HTS' version '2.11'
Building Bio-DB-HTS
Error: no compiler detected to compile 'lib/Bio/DB/HTS.c'. Aborting
ERROR: Shared Bio::DB:HTS library not found
解决办法:
cpanm Module::Build
cd /data/software
git clone https://github.com/Ensembl/Bio-DB-HTS.git
cd Bio-DB-HTS
#perl -MCPAN -e 'install Bio::Perl' # 提示么有bioperl
报错4:
(nextNEOpi) [sam@c01 ensembl-vep]$ conda install -c bioconda ensembl-vep
Collecting package metadata (repodata.json): done
Solving environment: failed with initial frozen solve. Retrying with flexible solve.
Solving environment: \
Found conflicts! Looking for incompatible packages.
This can take several minutes. Press CTRL-C to abort.
failed
UnsatisfiableError: The following specifications were found to be incompatible with each other:
Output in format: Requested package -> Available versions
Package libgcc-ng conflicts for:
conda-forge::python=3.6.10 -> libgcc-ng[version='>=7.3.0']
conda-forge::python=3.6.10 -> libffi[version='>=3.2.1,<3.3.0a0'] -> libgcc-ng[version='>=11.2.0|>=7.2.0|>=7.5.0|>=12|>=9.4.0']
Package zlib conflicts for:
conda-forge::python=3.6.10 -> zlib[version='>=1.2.11,<1.3.0a0']
conda-forge::python=3.6.10 -> sqlite[version='>=3.30.1,<4.0a0'] -> zlib[version='>=1.2.12,<1.3.0a0|>=1.2.13,<1.3.0a0|>=1.2.13,<2.0a0']
Package xz conflicts for:
ensembl-vep=106 -> htslib -> xz[version='5.2.*|>=5.2.3,<5.3.0a0|>=5.2.4,<5.3.0a0|>=5.2.5,<5.3.0a0|>=5.2.6,<5.3.0a0|>=5.2.5,<6.0a0']
conda-forge::python=3.6.10 -> xz[version='>=5.2.5,<5.3.0a0']
Package libstdcxx-ng conflicts for:
conda-forge::python=3.6.10 -> libstdcxx-ng[version='>=7.3.0']
conda-forge::python=3.6.10 -> libffi[version='>=3.2.1,<3.3.0a0'] -> libstdcxx-ng[version='>=7.2.0']The following specifications were found to be incompatible with your system:
- feature:/linux-64::__glibc==2.28=0
- feature:|@/linux-64::__glibc==2.28=0
Your installed version is: 2.28
报错5
注释的时候,提示没有插件库
singularity exec /home/sam/project/neoantigen/lib/nextNEOpi/nextNEOpi/singularity/pVACtools_3.0.2_icbi_3d50ac02.sif pvacseq generate_protein_fasta -p sample1_vep_phased.vcf.gz -s sample1_tumor sample1_hc_vep_pick.vcf.gz 31 sample1_long_peptideSeq.fasta
Command error:
VCF doesn't contain VEP FrameshiftSequence annotations. Please re-annotate the VCF with VEP and the Wildtype and Frameshift plugins.
需要安装对应的库:
singularity exec /home/sam/project/neoantigen/lib/nextNEOpi/nextNEOpi/singularity/ensembl-vep:106.1--pl5321h4a94de4_0 vep_install -a p -c /home/sam/project/neoantigen/lib/nextNEOpi/nextNEOpi/resources/databases/vep_cache --PLUGINS all 2
Available plugins: AncestralAllele,Blosum62,CADD,CAPICE,CSN,Carol,ClinPred,Condel,Conservation,DisGeNET,Downstream,Draw,ExAC,ExACpLI,FATHMM,FATHMM_MKL,FlagLRG,FunMotifs,G2P,GO,GeneSplicer,Gwava,HGVSIntronOffset,IntAct,LD,LOEUF,LOVD,LoF,LoFtool,LocalID,MPC,MTR,Mastermind,MaxEntScan,NMD,NearestExonJB,NearestGene,PON_P2,Phenotypes,PostGAP,PrimateAI,ProteinSeqs,REVEL,ReferenceQuality,SameCodon,SingleLetterAA,SpliceAI,SpliceRegion,StructuralVariantOverlap,SubsetVCF,TSSDistance,dbNSFP,dbscSNV,gnomADc,miRNA,neXtProt,satMutMPRA
Wildtype and Frameshift plugins 需要手动下载: https://pvactools.readthedocs.io/en/latest/pvacseq/input_file_prep/vep.html#vep
参考资料
- https://www.jianshu.com/p/1d43976aad78
- https://mp.weixin.qq.com/s/rjgSfzNK31t59FEmCKOUzA
- https://www.jianshu.com/p/572dde7176a3
- https://mp.weixin.qq.com/s/rjgSfzNK31t59FEmCKOUzA
- https://www.ensembl.org/info/docs/tools/vep/script/vep_download.html
- https://asia.ensembl.org/info/genome/variation/prediction/predicted_data.html#consequences
