【2.3】一代测序文件--ABI
二、其他
2.1 绘制ABI痕迹
ABI跟踪文件可用于检查。 虽然有现成的解决方案,但您可能会发现自己需要编写自己的代码以编程方式与跟踪交互。 Biopython允许我们这样做。
我在下面展示了一个交互式的IPython终端会话,使用我自己的Sanger测序文件,但它可以与其他AB1文件一起使用。
首先,我们在ABI文件中读到:
In [2]: from Bio import SeqIO
In [6]: record = SeqIO.read('55-Mn-fw-EM-28.ab1', 'abi')
我们最感兴趣的数据是记录的注释属性。
In [7]: record.annotations.keys()
Out[7]: dict_keys(['dye', 'abif_raw', 'sample_well', 'run_finish', 'machine_model', 'run_start', 'polymer'])
abif_raw下是另一个数据字典。
In [9]: record.annotations['abif_raw'].keys()
Out[9]: dict_keys(['DATA5', 'DATA8', 'RUNT1', 'phAR1', 'DATA4', 'DATA3', 'RUNT4', 'LsrP1', 'ASPt2', 'CTNM1', 'RUND1', 'RPrN1', 'DATA10', 'InSc1', 'SPAC2', 'phCH1', 'LNTD1', 'phQL1', 'S/N%1', 'RUND2', 'APrV1', 'SVER2', 'HCFG2', 'DyeW3', 'PLOC2', 'P1AM1', 'PTYP1', 'PDMF1', 'FVoc1', 'FTab1', 'RMXV1', 'TUBE1', 'DATA11', 'Tmpr1', 'BCTS1', 'SCAN1', 'EVNT1', 'DATA12', 'RMdX1', 'PBAS1', 'APXV1', 'User1', 'CpEP1', 'EVNT2', 'P2BA1', 'DATA1', 'APrN1', 'RUNT3', 'DySN1', 'B1Pt1', 'CTTL1', 'SVER3', 'SPAC1', 'APrX1', 'DATA9', 'RGNm1', 'EVNT4', 'RUND3', 'RUNT2', 'MODL1', 'PCON1', 'GTyp1', 'DyeW4', 'phTR2', 'PCON2', 'P2AM1', 'CTID1', 'CTOw1', 'ASPF1', 'EPVt1', 'SPAC3', 'SMLt1', 'NOIS1', 'NAVG1', 'B1Pt2', 'MCHN1', 'Rate1', 'DyeN2', 'AUDT1', 'RunN1', 'CMNT1', 'DSam1', 'DyeN3', 'RMdN1', 'AEPt1', 'RMdV1', 'P1WD1', 'Scan1', 'Scal1', 'InVt1', 'DyeN1', 'HCFG4', 'HCFG3', 'EVNT3', 'LIMS1', 'RUND4', 'ASPt1', 'SMED1', 'DyeW1', 'DCHT1', 'LAST1', 'PXLB1', 'SMPL1', 'Dye#1', 'APFN2', 'LANE1', 'P1RL1', 'P2RL1', 'FWO_1', 'RPrV1', 'phDY1', 'SVER1', 'DATA2', 'HCFG1', 'NLNE1', 'PLOC1', 'phTR1', 'RGOw1', 'DyeW2', 'Feat1', 'ARTN1', 'PSZE1', 'BufT1', 'DATA7', 'PBAS2', 'DyeN4', 'MODF1', 'AEPt2', 'PDMF2', 'DATA6'])
根据ABI数据规范(第40页),传统显示的跟踪所需的所有数据都在DATA9到DATA12通道中。 我们可以通过编程方式获取这些频道。 但是,没有明确说明,哪些字母对应于哪种颜色,因此对应于哪个确切的通道。
In [10]: channels = ['DATA9', 'DATA10', 'DATA11', 'DATA12']
In [13]: from collections import defaultdict
In [15]: trace = defaultdict(list)
In [16]: for c in channels:
...: trace[c] = record.annotations['abif_raw'][c]
...:
现在,可以在matplotlib图上绘制它们。
In [25]: plt.plot(trace['DATA9'], color='blue')
Out[25]: [<matplotlib.lines.Line2D at 0x10c956da0>]
In [26]: plt.plot(trace['DATA10'], color='red')
Out[26]: [<matplotlib.lines.Line2D at 0x10c8d7ac8>]
In [27]: plt.plot(trace['DATA11'], color='green')
Out[27]: [<matplotlib.lines.Line2D at 0x10c8f7908>]
In [28]: plt.plot(trace['DATA12'], color='yellow')
Out[28]: [<matplotlib.lines.Line2D at 0x10c96e128>]
In [29]: plt.show()
在放大特定区域后,我们将获得以下跟踪。
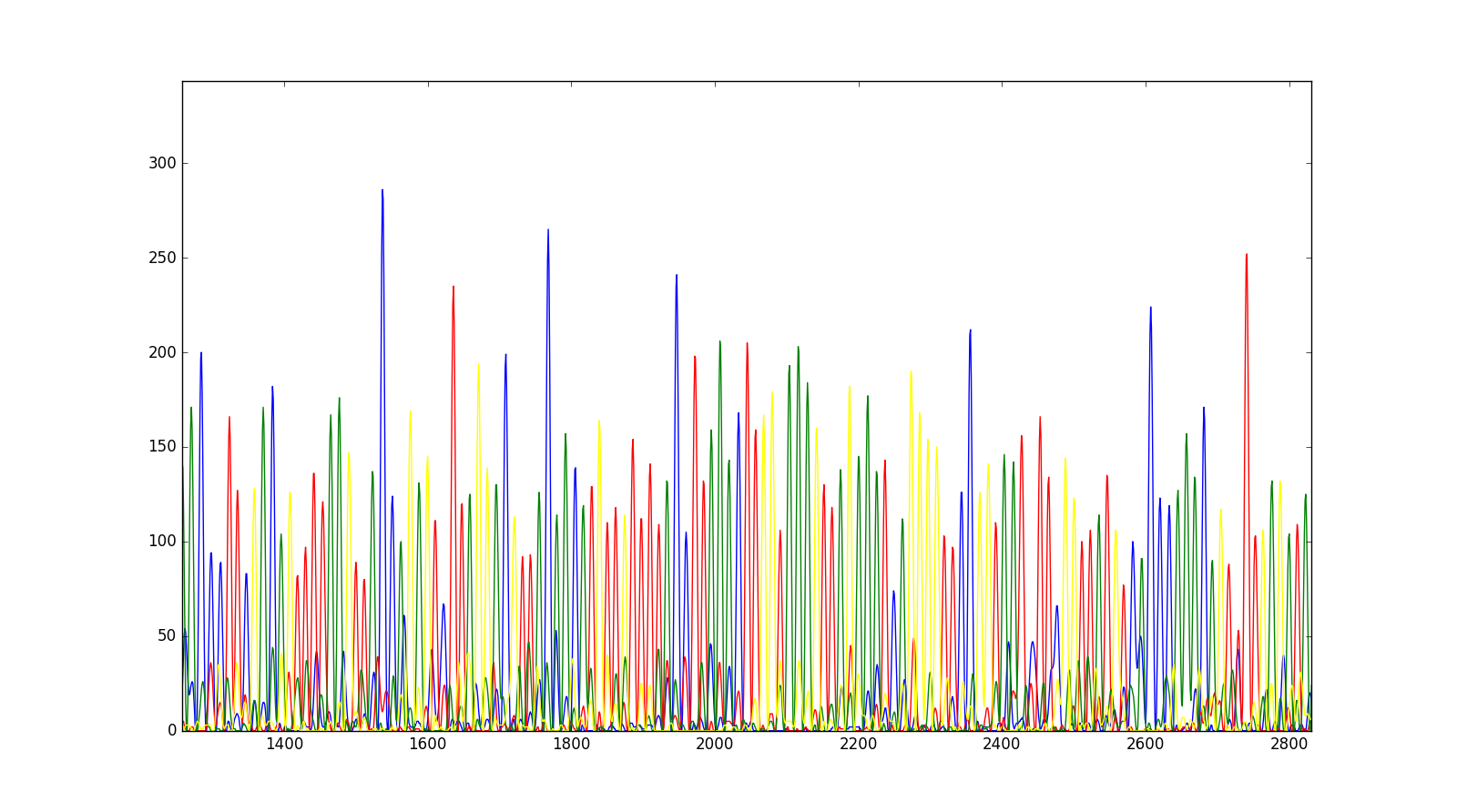
- 截至目前的写作,这本食谱配方没有进一步深入计算指标。 可能有趣的一个特别的事情是量化混合碱基调用与单碱基调用的Shannon多样性。
- 另外需要注意的是:数据中每个碱基有10个色谱图值。 因此,这意味着建议将每个第5个值取出,以便序列跟踪数组的最终长度与应该排序的位置数相匹配,而不是大10倍。(????)
其他工具
CutePeaks

https://github.com/labsquare/CutePeaks
参考资料
药企,独角兽,苏州。团队长期招人,感兴趣的都可以发邮件聊聊:tiehan@sina.cn
![]() 个人公众号,比较懒,很少更新,可以在上面提问题,如果回复不及时,可发邮件给我: tiehan@sina.cn
个人公众号,比较懒,很少更新,可以在上面提问题,如果回复不及时,可发邮件给我: tiehan@sina.cn
