【3.5.1.1】gtf格式
提供基因位置的注释文件通常以GTF(General Transfer Format)或GFF3(General Feature Format)格式呈现。有GTF文件后,就可以利用注释信息计算每个基因/转录本/外显子比对了多少reads,从而获取counts值。GTF是GFF的便于传输版。
GTF格式各列的内容为:
seqname - 染色体或scaffold的名称。
source - 生成这个特征的项目名称,或数据库来源。
feature - 特征类型名称,如gene、transcript、exon、CDS。
start - 开始位置,使用基于1的索引。
end - 结束位置,使用基于1的索引。
score - 组装的转录本的可信度分数(目前该字段未被StringTie使用,如果转录本与read alignment bundle有连接,StringTie将报告一个常量值1000)。
strand - 正链或负链+/-。
frame - 密码子的第几个碱基0/1/2(StringTie不使用这个字段,只记录一个点.)。
attribute - 附加信息。
例如:
a. 来自ensembl的果蝇的Drosophila_melanogaster.BDGP6.32.109.gtf。
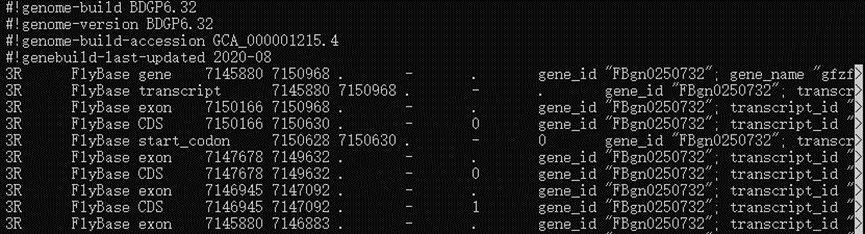
feature 的内容是gene、transcript、exon、start_codon、stop_codon、five_prime_utr、three_prime_utr、CDS、Selenocysteine。 attributes的内容例如:gene_id “FBgn0250732”; transcript_id “FBtr0091512”; gene_name “gfzf”; gene_source “FlyBase”; gene_biotype “protein_coding”; transcript_name “gfzf-RB”; transcript_source “FlyBase”; transcript_biotype “protein_coding”; tag “Ensembl_canonical”;
b. 来源UCSC的人类GTF文件,1号染色体是 chr1。

c. 来源Ensembl的Homo_sapiens.GRCh38.chr.gtf.gz,1号染色体是1。

d. Stringtie得到的sample.gtf。
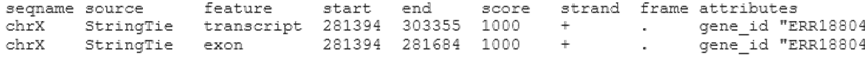
Stringtie得到的sample.gtf的attributes是以分号分隔的tag-value pairs列表,包括:gene_id、transcript_id、exon_number、reference_id、ref_gene_id、ref_gene_name、cov(转录本或外显子的平均每碱基覆盖率)、FPKM、TPM。例如:gene_id “ERR188044.1”; transcript_id “ERR188044.1.1”; reference_id “NM_018390”; ref_gene_id “NM_018390”; ref_gene_name “PLCXD1”; cov “101.256691”; FPKM “530.078918”; TPM “705.667908”
GFF格式各列的内容为:
seqid - 染色体或scaffold的名称。
source - 生成这个特征的项目名称,或数据库来源。
feature - 特征类型名称,来自SOFA sequence ontology。
start
end
score
strand - 正链或负链+/-。
phase - 密码子的第几个碱基0/1/2。
attribute - 附加信息。A semicolon-separated list of tag-value pairs。
GTF和GFF之间的区别:
- 数据结构:都是由9列构成,分别是reference sequence name; annotation source; feature type; start coordinate; end coordinate; score; strand; frame; attributes.前8列都是相同的,第9列不同。
- GFF第9列:都是以键值对的形式,键值之间用“=”连接,不同属性之间用“;”分隔,都是以ID这个属性开始。
- GTF第9列:同样以键值对的形式,键值之间是以空格区分,值用双引号括起来;不同属性之间用“;”分隔;开头必须是gene_id, transcript_id两个属性。
参考资料
