【2.1.5.1】VMD看amber结果
本教程旨在向您介绍如何将VMD与AMBER一起使用,介绍如何加载AMBER轨迹文件和inpcrd文件以及如何处理数据。它并非旨在全面涵盖VMD中存在的所有许多功能。
一、介绍
可视分子动力学(VMD),可从 http://www.ks.uiuc.edu/Research/vmd/ 获得,它是一种功能强大且功能丰富的分子可视化程序包,可使用3-D显示图形和内置脚本,设置动画和分析大型生物分子系统。它的发展由国家卫生研究院(National Institute of Health)在克劳斯·舒尔滕(Klaus Schulten)的指导下提供资金。它对学术研究人员免费提供,注册后可以从上面的链接下载。
VMD是用于可视化AMBER软件套件的pmemd模块产生的结果的理想工具。但是,它的使用在其他地方并不直观。因此,设计本教程的目的是简要介绍如何可视化和操纵从AMBER仿真产生的轨迹。
本教程基于VMD v1.8.3,在撰写本文时(2005年6月3日)是最新版本。已针对VMD v1.9.2更新。如果使用其他版本的VMD,则可能会发现某些功能不可用或界面略有不同。但是,仍然有可能完成本教程的绝大部分。
本教程还假定您正在使用Linux。如果您的操作系统不同,则某些布局和/或过程可能会略有不同。
本教程包括九个部分:
- section1.htm:加载VMD和自定义初始窗口布局
- section2.htm:加载PDB文件
- section3.htm:更改表示形式
- section4.htm:加载AMBER inpcrd和restrt文件
- section5.htm:对准分子并测量RMSD
- section6.htm:可视化amber轨迹
- section7.htm:保存单个坐标集
- section8.htm:在轨迹过程中跟踪系统参数
- section9.htm:制作电影
二、常用的操作
2.1 加载VMD和自定义初始窗口布局
在我们开始研究一些AMBER轨迹之前,您应该确保VMD已正确安装在您的计算机上并且可以正常工作。 发出命令“ VMD”时,它应该加载而不会出现错误消息。 如果无法使VMD正常工作,请访问VMD网站以获取更多信息。
让我们从自定义VMD的启动方式开始,以便我们拥有相似的显示布局。 (如果您有信心在VMD上导航,则可以跳过此步骤。)
由于大多数人将使用单个显示器来运行本教程,因此我们将逐步为单个显示器创建启动脚本。
VMD的初始布局由名为.vmdrc的文件控制。 这是一个 .vmdrc 文件,您可以使用它来获取我认为是良好的初始布局。
vmdrc
# turn on lights 0 and 1
light 0 on
light 1 on
light 2 off
light 3 off
# position the stage and axes
axes location off
stage location off
# position and turn on menus
menu main on
menu graphics on
menu files on
menu main move 5 196
menu graphics move 5 455
menu files move 800 750
display projection orthographic
如果您的主目录中有一个名为.vmdrc的文件(请使用cd〜,ls -la检查),请将其删除或重命名。然后将上面的文件另存为您的主目录中的“ .vmdrc”。 (请注意vmdrc之前的点)。
该文件由VMD在加载时自动获取,并执行多项操作。首先,它打开灯0和1。然后关闭轴。我觉得这会创建一个更清洁的OpenGL窗口。它还将显示投影模式设置为正交,在我看来,当在没有精确的4:3比例的屏幕上旋转时,例如,在屏幕上旋转时,可以提供更好的深度感知和更少的失真。我的屏幕分辨率为1280x1024。最后,它打开主菜单,图形菜单和文件菜单。然后将它们移动到屏幕上合适的位置。注意:这是为1280x1024的屏幕分辨率设计的。如果分辨率不同,则可能需要使用3条移动线上的数字来获得合理的布局。
现在运行VMD,它应该具有类似于以下内容的布局:
>vmd
我们现在准备尝试加载一些分子。
2.2 载入PDB文件
可以从蛋白质数据库中获取的PDB文件,可以通过以下两种方式之一加载到VMD中。 您可以在命令行中指定它们,例如:
vmd my_structure.pdb
或者,您可以通过“分子文件浏览器”加载它们。 让我们尝试使用后一种情况加载肝脏酒精脱氢酶(LADH)。 这是LADH的pdb( 1LDY.pdb )。 注意:您可能需要右键单击此链接,然后选择另存为。 如果尚未启动,请通过单击“文件”->“新分子”打开“分子文件浏览器”窗口。
然后选择浏览并查找您刚刚保存的1LDY.pdb文件。 接下来,选中“确定文件类型”框以确保其显示为PDB。 永远不要信任VMD来获得正确的结果,尤其是在加载AMBER轨迹时。 然后点击Load,然后弹出四聚体LADH:

默认情况下,鼠标将处于“旋转”模式。 在鼠标指针位于“ Open GL”显示屏内的同时按住鼠标左键,可以旋转分子。 鼠标还有两种其他模式:“平移”(使分子在窗口平面内移动)和“缩放”(Scale)使您可以放大和缩小分子。 您可以通过两种方式访问这些模式。 第一种是通过鼠标菜单:

第二种方法(可能是更方便的方法)是使用“鼠标菜单”右侧显示的键盘快捷键:
(r)=旋转模式 (t)=Translate Mode (s)=缩放模式
当鼠标悬停在“ OpenGL”窗口上方时,按“ s”,然后在按住鼠标左键的同时左右移动鼠标。 按“ r”返回旋转模式。 如果有任何麻烦,请单击“显示”->“重置视图”以返回到默认视图。
2.3 更改表示形式
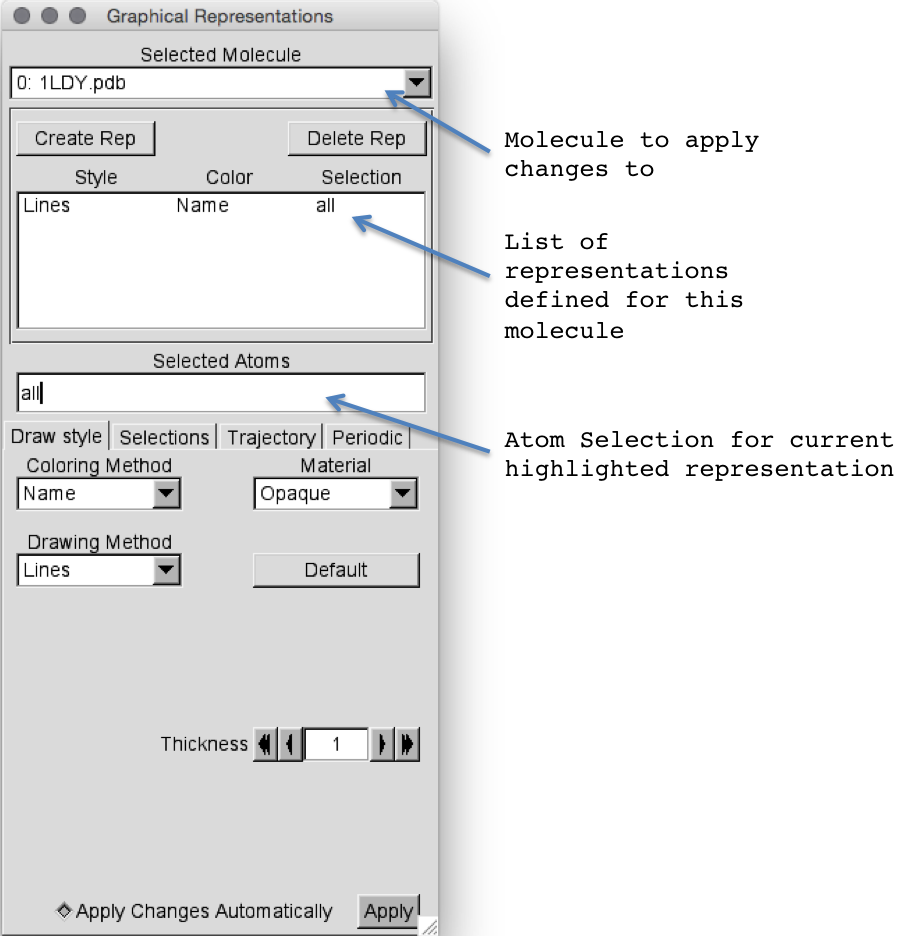
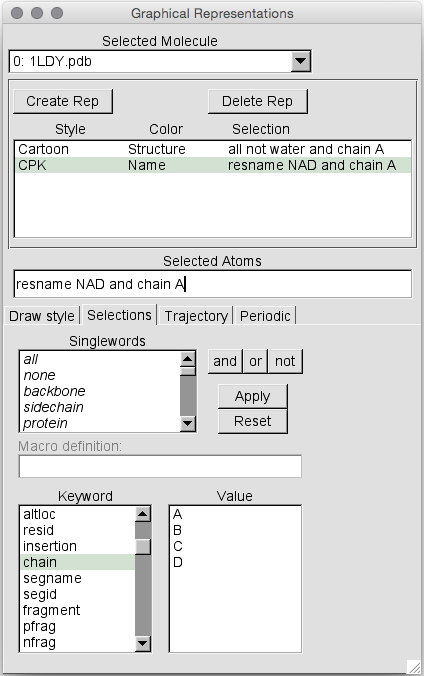
现在我们已经掌握了操作分子视图的基础知识,接下来要看的是如何更改表示形式。 这是通过“图形表示”窗口完成的。 如果尚未打开,请单击“图形”->“表示”以将其打开。
默认情况下,“选定分子”将是您打开的最后一个。 在这种情况下,分子0:1LDY.pdb(注意:VMD从零开始。这也适用于残基,例如残基0实际上是残基1 !!!)。 通过单击所选分子旁边的向下箭头,您可以更改我们要应用更改的分子。 在这种情况下,我们仅加载一个分子,因此它是默认值。
让我们先从显示屏上去除水域。 如果您现在看显示屏,将会看到所有这些小红点。 这些是pdb文件中结晶水的氧原子。 我们可以通过更改选定原子框将它们从当前表示中删除:
将所选原子框中的“all”替换为“all not water”,然后单击“应用”。 红点应消失。 尽管此示例可能看起来并不惊人,但在查看显式溶剂计算时却非常有用,因为水分子常常会使您感兴趣的蛋白质或系统模糊。 接下来,我们将视图更改为卡通视图,以便我们可以更清晰地看到结构。
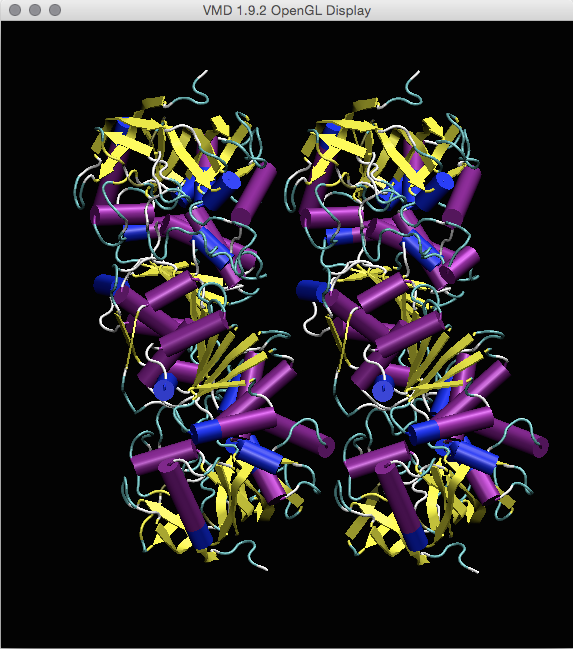
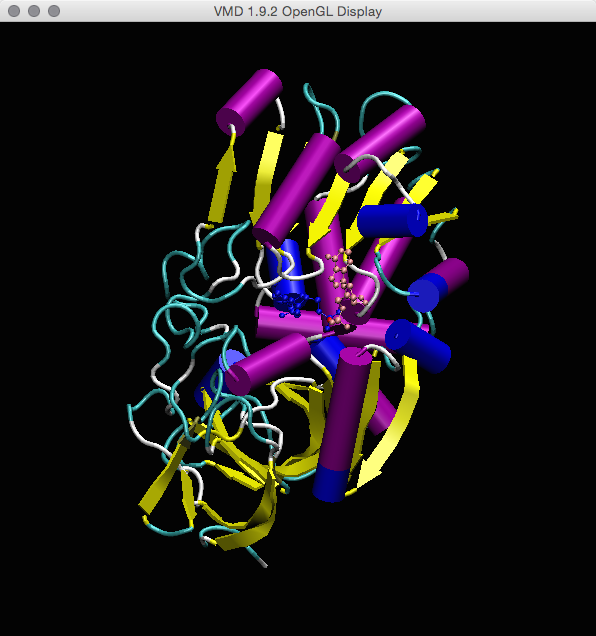
在Coloring Method中,选择“Secondary Structure”。 这将使颜色变成一种颜色,其中不同的残基将根据其所属的二级结构而具有不同的颜色。 接下来,从“Drawing Method”框中选择“Cartoon”。 (如果您使用的是v1.8.3或更高版本,也可以尝试“ NewCartoon”)。 现在,您的显示应如下所示:
接下来,我们将突出显示此蛋白质中的NADH辅酶分子,以便我们可以看到它们。 为此,我们将为此分子创建第二个表示形式,并仅选择名称为NAD的那些残基。 因此,在“Graphical Representations”窗口中,单击“Create Rep”,您将获得另一个与原始表示相同的表示。 接下来单击选择选项卡:
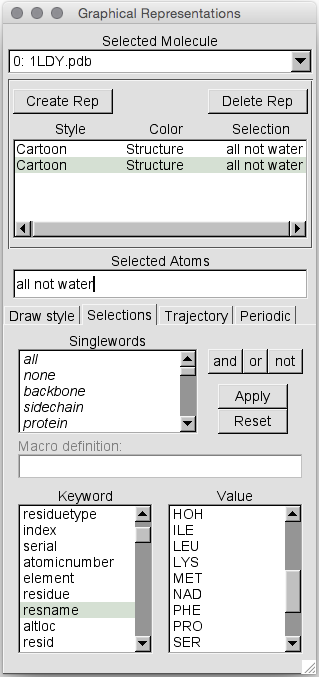
在这里,我们可以在“Selected Atoms”框中选择所需的选择。 注意,如果我们知道想要什么,我们也可以像在去除水的过程中一样简单地在此框中键入内容。 因此,让我们仅选择名称为“ NAD”的残基。 首先,删除“Selected Atoms”框中的文本。 然后双击resname(这会将resname添加到“所选原子”文本框中)。 然后向下滚动值列表,然后双击NAD。 然后点击“应用”。 现在我们可以回到“绘制样式”选项卡:
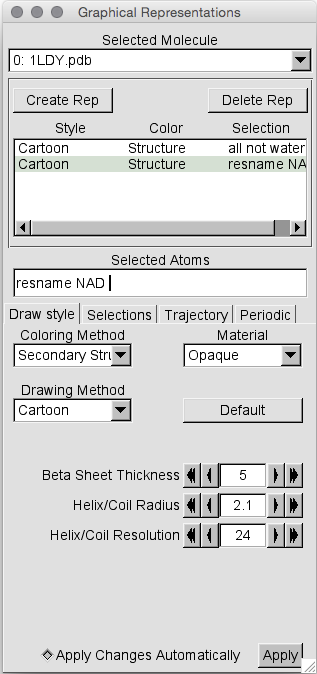
就目前而言,我们分子的表示并未发生多大变化。 这是因为Draw样式仍设置为Cartoon。 让我们将其更改为CPK,以便可以看到NADH残基。 第1步:确保两个表示形式中的第二个都如上所述突出显示-我们不想将整个蛋白质更改为CPK,因为这确实会伤害眼睛(如果您不相信我,请尝试一下)…
接下来,将“Coloring Method”更改为“Name”,将“Drawing Method”更改为CPK。 然后点击“应用”。 您现在应该可以看到4个NADH残基。 因此,我们有一个四聚体。 好吧,实际上我们附近有两个二聚体。 如果旋转蛋白质,您将可以看到两个二聚体之间显然存在溶剂填充的空隙。 您可以通过双击制图表达列表中的名称来暂时关闭这两种制图表达:

尝试一下,双击选择“all not water”的那个。 您应该只剩下NADH残基。
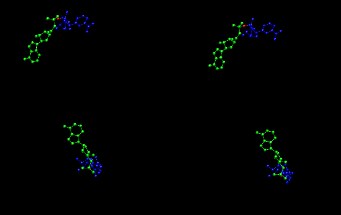
再次双击它,您将得到蛋白质。
现在:看看您是否可以算出如何仅显示具有单个NADH残基的蛋白质的链A:
提示:您将需要同时修改两个表示形式的选定原子。 您还将需要’and’关键字。
2.4 加载AMBER rst7和restrt文件
让我们删除当前分子,然后尝试查看xleap或sander创建的AMBER rst7或restrt文件。 首先去除LADH分子。 在“ VMD Main”窗口中,右键单击1LDY.pdb文件名,然后选择“ Delete Molecule”。 您可能必须先左键单击以选择它。
接下来,我们将打开我使用xleap创建的prmtop和rst7文件。不用担心这些文件是如何创建的。这将在以后的教程中介绍。这是您将需要的两个文件:TRPcage.prmtop,TRPcage.rst7。
加载AMBER结构与加载pdb文件的方式略有不同,因为实际上有两个文件。第一个文件是prmtop或拓扑文件。这与分子中原子的位置无关,它仅定义了每个原子是什么,与原子键合的对象以及每种原子类型的参数。另一方面,rst7文件仅列出了一组坐标,但没有提及原子。因此,我们需要这两个文件来加载结构。让我们先看一下pdb视图的局限性之一。这是我使用ambpdb创建的TRPcage结构的pdb。 (TRPcage.pdb)。
我使用xleap的sequence命令手动创建了这种结构,因此它的质子和残基侧链位置非常差。将pdb加载到VMD中,然后查看得到的内容。它看起来应该不错。但是,请尝试放大中央色氨酸残基。你看到了什么?
如果除去其他残基,可能会更容易了解发生了什么。在“Selected Atoms”下的“Graphical Representations”中,输入“all within 5 of resname TRP”。这只会显示位于称为TRP的残基5埃内的原子。在那里,还要选择CPK作为绘图方法。接下来,在“ VMD Main”窗口中单击“ Mouse”菜单,然后单击“ center”。这使我们可以更改旋转中心。现在单击tryptophan的backbone nitrogen 。现在,当您旋转时,它应该围绕此原子旋转。 (按r返回旋转模式)。
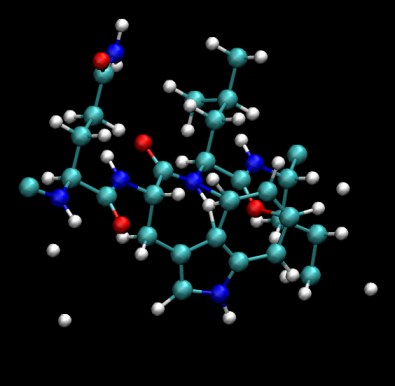
那些奇怪的bonds是什么?我们有一个带有3个键的质子。当然,模拟不是以这种方式设置的。确实不是。但是我创建的pdb中没有bond信息。这样,VMD根据原子之间的距离添加了键。由于这是一个“非常”糟糕的起始结构,因此有许多原子非常紧密地结合在一起,因此VMD将它们全部结合在一起。因此,查看pdb文件不会告诉您Amber将使用的bond在哪里。为了查看Amber如何处理该分子的键合,我们必须使用prmtop文件。
因此,删除此分子,以便我们可以加载一个prmtop。加载AMBER prmtop文件时,重要的是要知道是拥有新格式的prmtop文件(使用AMBER7或更高版本创建)还是旧的文件(使用AMBER 6或更早版本创建)。我在上面给您提供的prmtop是一种新格式。因此,转到分子文件浏览器并浏览prmtop文件。现在,在“Determine file type”下,选择“ parm7” [vmd1.8.3]或“ AMBER 7 Parm” [vmd1.8.4及更高版本]。这表示新的prmtop格式(parm表示旧格式)。点击加载。
发生了什么事吧?这是因为prmtop文件不包含坐标。但是,它应该已经创建了一个名为“ TRPcage.prmtop”的新结构容器。现在,通过确保在Molecule File Browser的“Load files for:”中选择了该分子,我们可以将任何数量的结构加载到该分子中。

现在我们可以或多或少地将任何分子格式加载到该分子中。 如果将pdb文件加载到该分子中,我们将获得与以前相同的键,但是这次的bond将是prmtop文件中定义的绑定。 试试吧。 我们还可以将rst7文件(由LEaP创建)或restrt文件(由sander创建)加载到该分子中。 为此,我们浏览文件(TRPcage.rst7),然后选择“ rst7” [vmd1.8.3]或“ AMBER7 Restart” [vmd1.8.4]作为文件类型。 确保在““Load files for:”框中选择了TRPcage.prmtop,然后单击“加载”。
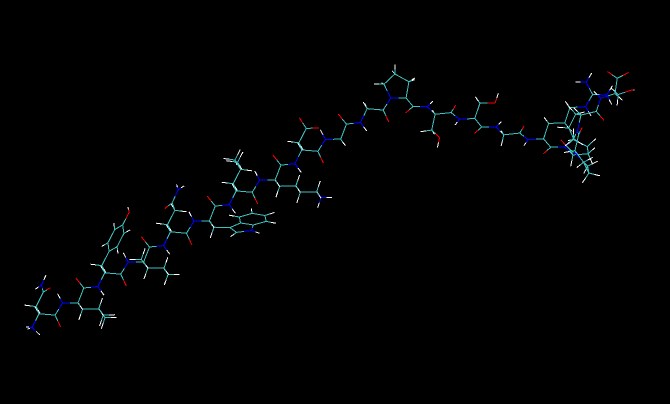
现在bond应该看起来好多了。但是,这仍然太过紧张,无法运行MD,因此我通过使用sander为您minimized了它。这是最小化结束时的restrt文件。 (TRPcage.ncrst)。
我们可以将其加载为新分子(通过再次加载prmtop文件)或作为当前分子中的新框架。我想同时看到两个分子,因此我将其作为新分子加载。因此,再次转到“Molecule File Browser”窗口,然后在“Load files for:”框中选择“New Molecule”。现在,单击“Browse”按钮并浏览以查找TRPcage.prmtop文件。选择“ AMBER7 Parm”作为类型,然后单击“加载”。现在,再次单击浏览,找到TRPcage.ncrst文件。选择“ AMBER7 Restart”作为类型,然后单击“加载”。现在,您应该在“ OpenGL”窗口中显示两个分子。他们将非常相似。为了查看它们之间的差异,让其中一个颜色为蓝色。
转到“Graphical Representation”窗口,然后在“Selected Molecule”下选择列表中两个中的第一个TRPcage.prmtop。这应该是我们的初始(最小化)结构。然后在“着色方法”下选择“ ColorID”,然后在其右侧的框中选择0。您应该看到“ OpenGL”窗口中的一个分子变为蓝色。现在,您可以放大此视图并查看两种结构之间的差异。
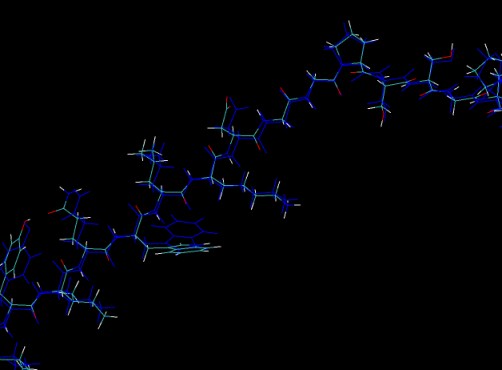
正如您可能已经猜到的那样,最小化已将色氨酸从主链上扭转开来,避免了我们之间真正不良的氢接触。 我们可以计算最小变化的RMSD吗? 我们可以使用AMBER的cpptraj命令执行此操作,但是也可以使用RMSD扩展名在VMD本身中执行此操作。
2.5 对准分子并测量RMSD
我们分两个阶段进行RMSD拟合。 首先,我们对齐两个分子,然后测量对齐的结构之间的RMSD。 对齐实际上涉及RMSD拟合,但实际值未打印到屏幕上。 为了说明对齐方式是如何工作的,首先让我们自己旋转一个分子。 为此,我们可以从第一个分子中删除“ Fixed”标志。 我们通过双击“ VMD Main”窗口中分子旁边的字母F来执行此操作。 当您双击它时,它应该变成黑色:
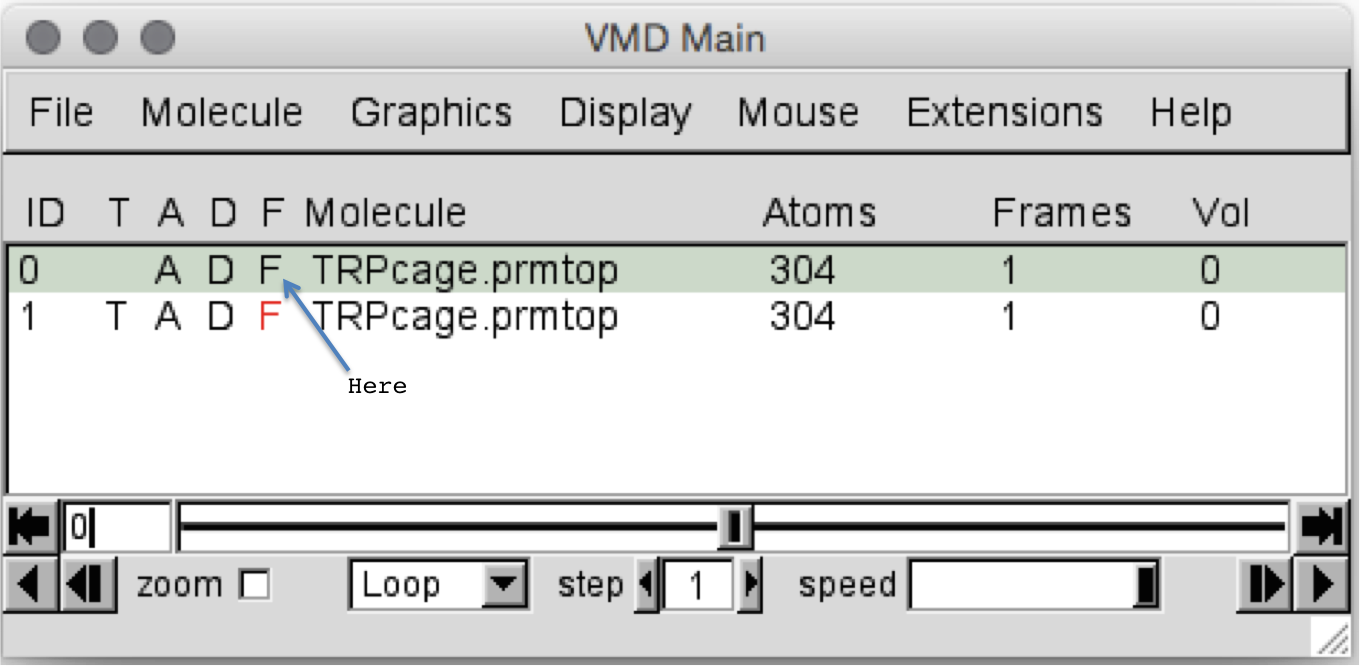
然后,当您返回“ OpenGL”窗口时,您应该发现只有第二个结构(我们最小化的一个)旋转。 现在,旋转分子,使其不再与第一个分子对齐,如果需要,您也可以对其进行平移:
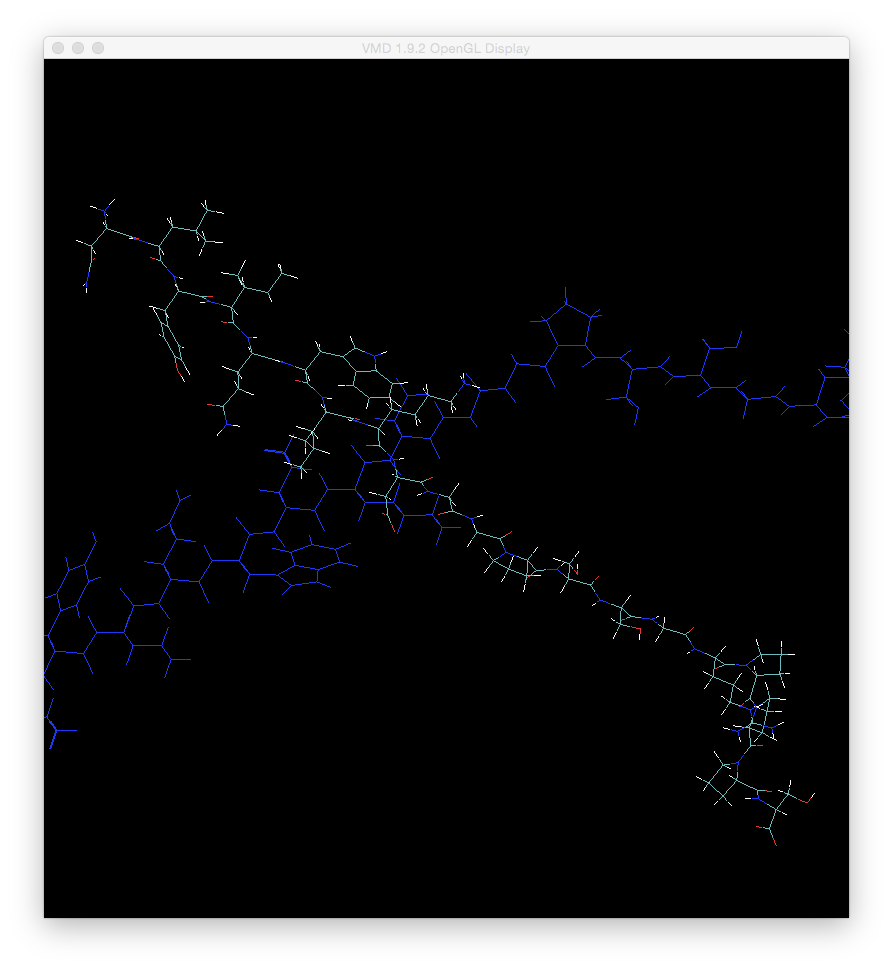
现在,再次双击第一个分子上的F标志,以使其不再固定。现在,您将发现这两个分子一起旋转,很难看到它们之间的结构差异。例如,如果加amber轨迹和晶体结构,通常会得到此结果。它们几乎永远不会具有相同的原点或轴定义,因此您无法直观地比较这两个分子。现在,我将向您展示对齐两个分子的快速方法。不幸的是,我们需要加载一个坐标已移位的新文件才能进行对齐(如果我们不这样做,只是尝试按照当前的方式进行对齐,VMD实际上不会重新对齐两个分子)。
这是我在x方向上偏移了2埃的结构(TRPcage_shifted.ncrst)。
现在,要确保我们位于同一位置,请退出VMD,然后重新加载它。
加载TRPcage.rst7结构。记住要先加载prmtop,选择AMBER7 Parm作为类型。然后选择“Load files for: New Molecule ”并加载prmtop,然后将TRPcage_shifted.ncrst结构加载为AMBER7 Restart。然后转到“Graphical Representations”,并将第一个分子(0:TRPcage.prmtop)的颜色更改为ColorID0。现在的显示应如下所示:
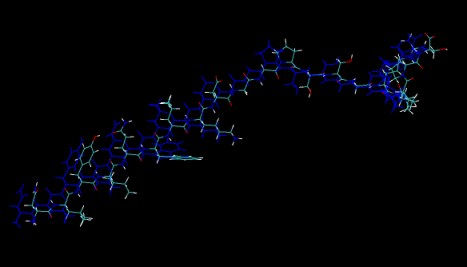
如您所见,这两个结构不再对齐,很难比较它们。
在VMD的早期,您必须使用AMBER的ptraj手动对齐两个分子,然后才能在VMD中打开它们。 确实,您仍然可以使用ctratraj(ptraj的更新版本)来执行此操作,并且在某些情况下(当您要做的事情非常复杂,或者希望将RMSD作为时间的函数时,等等),您将需要使用 cpptraj。 但是,通常您只是想进行快速比较,因此可以使用VMD的RMSD计算器。 要访问此选项,请单击Extensions->Analysis->RMSD Calculator。 请注意,这是一个部分,在各个版本之间进行了很大的更改,因此您的选择可能与我的略有不同:
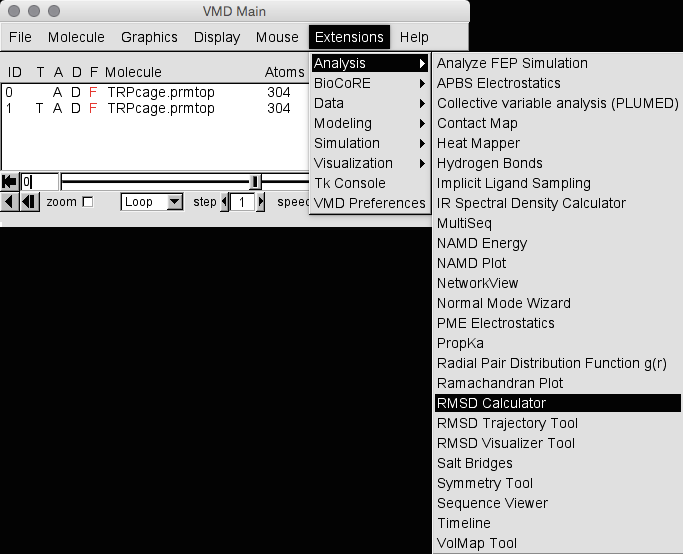
这将打开以下窗口:
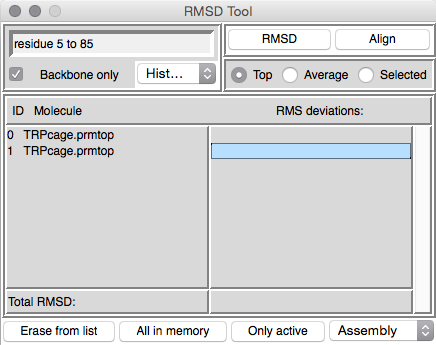
我们需要做的第一件事是在文本框中输入我们要填充的残基。比对将在整个分子上进行,但实际计算将根据我们指定的内容进行。在这种情况下,我们要适合所有残基,因此在窗口顶部的文本框中输入“ all”。接下来,我们只希望针对骨架原子(C,CA和N)进行RMS拟合,因此确保选择“Backbone only”。接下来,我们需要确保选择了正确的分子。我将首先点击“Erase from list”按钮,直到所有分子从列表中删除为止。下一个命中“All in memory”。这将添加我们当前已加载的两个分子。接下来,we need to select what we want to fit and what we want it to fit to。因为这里我们只有两个结构,所以这确实是很容易的,但是即使如此,我们还是将第二个结构(最小化的结构)fit第一个结构。点击选定 Selected tab,这应该取消选择顶部。接下来,单击分子列表中的第一个分子以将其选中:
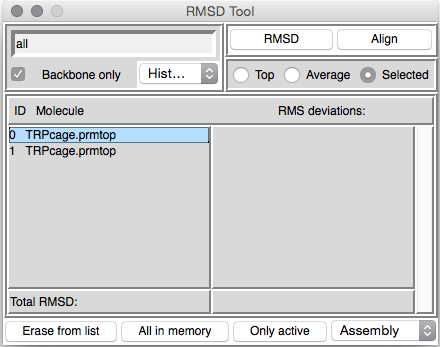
现在,如果我们此时点击RMSD,我们将获得两个结构之间的RMSD,目前,当我们旋转和平移其中一个结构时,它们是非常不同的。 尝试一下,按一下RMSD按钮。 您应该达到2.2埃左右。 这是因为一个偏移了另一个2埃。 现在单击“对齐”按钮。 您应该看到两个分子在“ OpenGL”窗口中对齐。 如果现在单击RMSD,您将在两个分子的主链原子之间获得正确的RMSD。 在这种情况下,约为0.57埃。 此值是最小化的典型值。
本教程的最后阶段是如何加载AMBER轨迹文件,以便我们可以观察结构随时间变化的“电影”。
2.6 可视化AMBER轨迹
注意:不幸的是,VMD尚无法加载压缩(压缩)的轨迹文件,因此,当您下载我在此处提供的nc.gz文件时,必须先解压缩然后使用gunzip,然后再将其加载到VMD中。
让我们首先加载一些轨迹文件,这些文件覆盖上面所示的TRPCage扩展结构的分子动力学模拟的前10ps。 这些模拟是使用Sander v8.0运行的。
以下是您需要的文件:TRPcage.prmtop(128 kb),heat1.nc.gz(510 kb),heat2.nc.gz(515 kb)
首先解压缩两个轨迹文件:
>gunzip heat*.nc.gz
如果VMD正在运行,请退出它。 这样,我们应该从同一点开始。 加载VMD并浏览以找到TRPcage.prmtop文件。 选择AMBER7 Parm,然后单击Load。 在这个结构中,我们将一个接一个地加载两个轨迹文件。 因此,加载第一个,浏览heat1.nc,然后选择“ AMBER Coordinates”作为类型并单击Load。
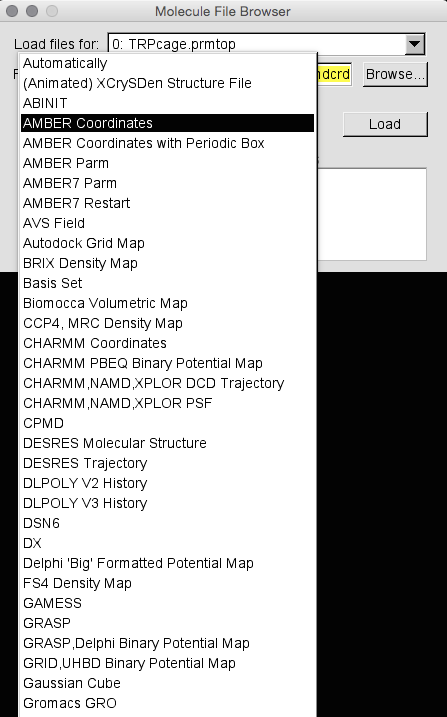
注意:有两种与amber轨迹(nc)文件相关的类型。 这些是“AMBER Coordinates”和“AMBER Coordinates with Periodic Box”。 两者之间的区别在于轨迹文件是来自气相/隐式溶剂/溶剂上限模拟(AMBER坐标)还是来自周期性模拟(AMBER Coordinates with Periodic Box)。 当sander从具有周期性边界的模拟运行中写入nc文件时,它会在每个帧的末尾写入额外的3个浮点数,它们是框的大小。 VMD不够智能,无法确定它是什么类型的轨迹文件,因此我们需要告诉它。 在这种情况下,我们需要进行隐式溶剂计算,因此请选择“AMBER Coordinates”。 您经常可以分辨出是否选择了错误的格式,因为看起来很奇怪。 随时尝试。 如果确实加载了看起来很奇怪的模拟,请尝试删除分子并重新加载,然后选择其他格式:
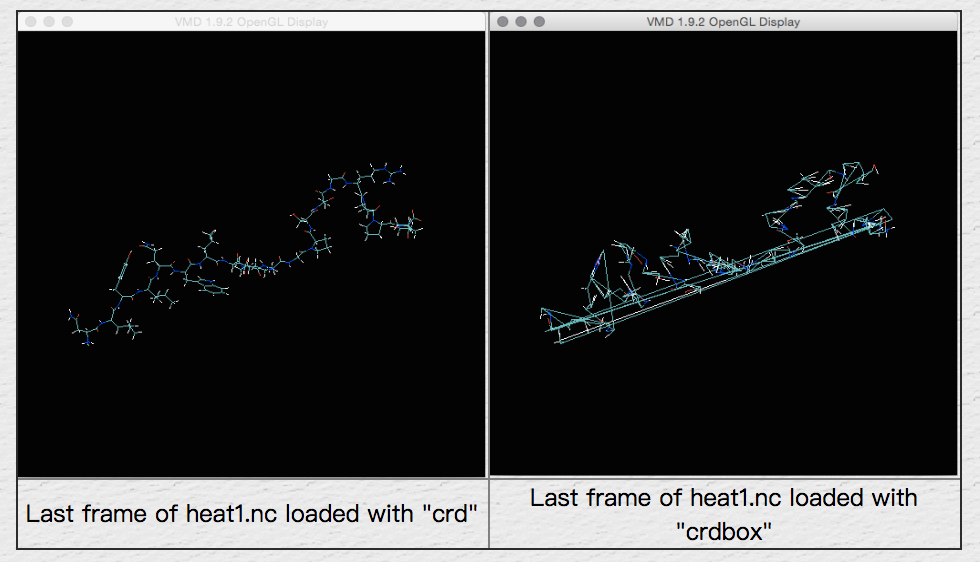
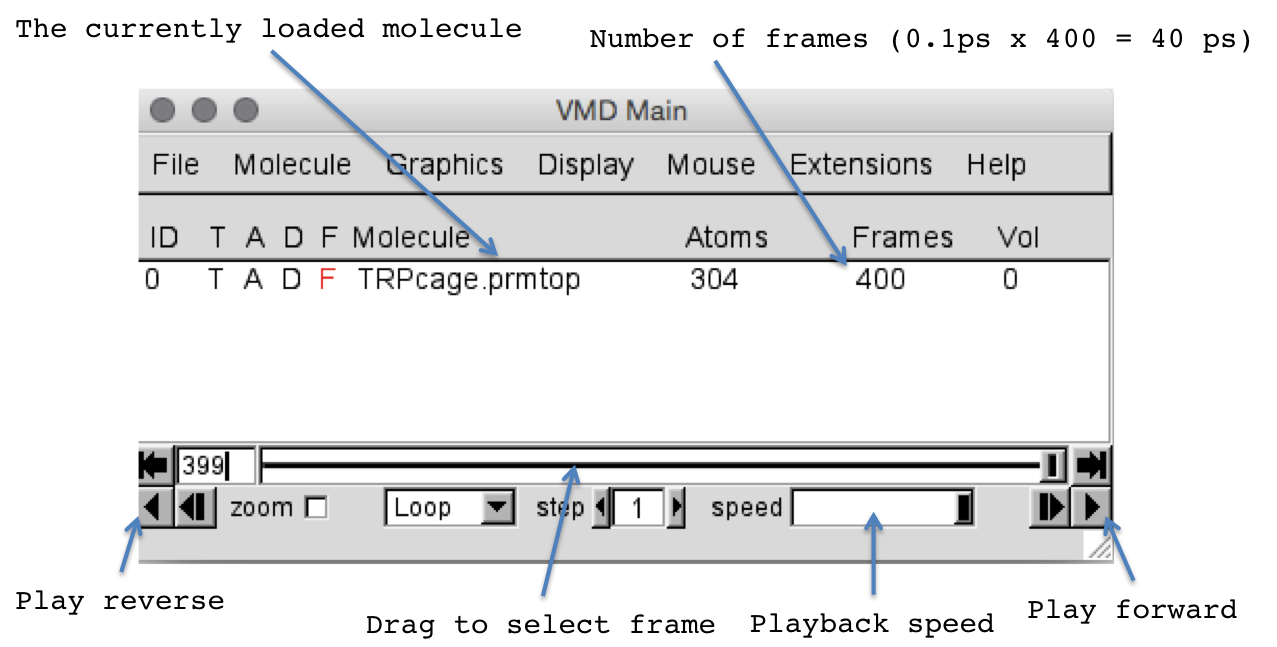
如果正确加载,则应该看到TRPcage结构出现在OpenGL窗口中并开始移动。 接下来,再次单击浏览,然后找到heat2.nc。 以相同的方式加载它(不需要重新加载prmtop文件)。 确保在“Load files for:”框中选择了TRPcage.prmtop。 这会将heat2中的帧添加到我们已经加载的帧中。 这样,您可以将多个不同的轨迹集加载到单个动画中,从而可以无间断地观看整个轨迹。 您现在应该已加载400 frames TRPcage。 从0K到100K,这涵盖了10ps的加热。 尝试重播轨迹。 您可以在“ VMD Main”窗口中使用影片控件:
2.7 保存一组坐标
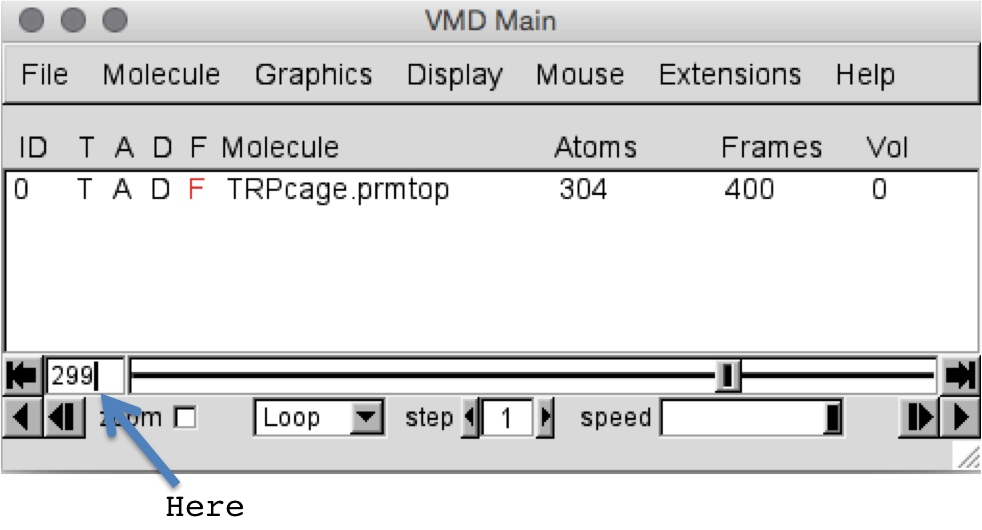
现在我们加载了轨迹,将帧300保存为pdb文件。 首先,我们必须确保在窗口中显示了第300帧。 VMD从零开始编号所有内容,因此实际上是“ VMD speak”中的第299帧。 通过在VMD主窗口中拖动栏旁边的文本框中键入frame号,我们可以轻松跳转到特定frame。 现在就试试。 输入299,如下所示,然后按Enter。
这就是我们要保存的结构。
接下来单击“Molecule”名称。具有T A D和F标志的文本行。 这应该以绿色突出显示该行。 然后单击File->Save Coordinates… .,将出现以下窗口:
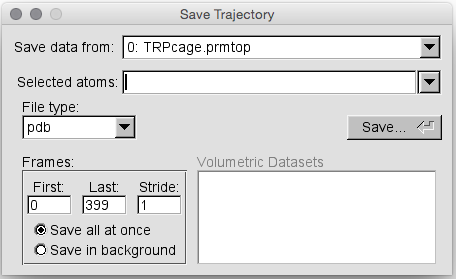
该工具实际上将使我们能够将整个轨迹另存为其他类型,例如 Binpos甚至pdb。 VMD会将轨迹的每一帧保存为一个新分子,保存在单个pdb文件中。 但是,如何解释这样的pdb文件似乎模棱两可。 某些程序(例如RasMol)会将所有分子加载在一起并覆盖它们,而其他程序(例如VMD)会将每个分子加载为轨迹中的框架。
出于我们的目的,我们只想将一个框架保存为pdb文件。 因此,首先在“选定的原子”框中键入all,这会将所有原子保存在我们的结构中。 接下来在文件类型下选择pdb。 最后,在“Frames box”中选择“第一帧( First frame )”为299,选择“最后一帧”为299:
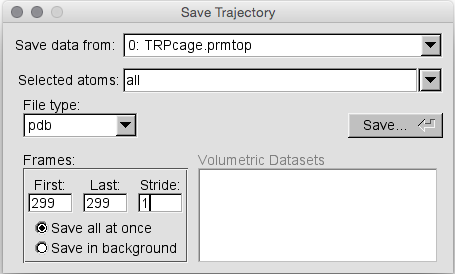
最后,点击保存并使用您选择的文件名保存文件。 这是您应该获得的pdb文件:( frame_300.pdb)。如果需要,可以随时将其重新加载到VMD中。
2.8 在轨迹过程中跟踪系统参数
在本教程中,我们还有两个有用的内容。 首先是使用VMD的标签工具来观察两个原子之间的距离在我们的轨迹中如何变化。
首先让我们当前加载的轨迹更漂亮。 单击Graphics->Representations 以调出“Graphical Representations”菜单(如果尚未启动)。 接下来,单击“Create Rep”,然后将 Drawing method更改为“Ribbons”,将Coloring method更改为“ ResType”。 然后再次点击“ Create Rep”,将 Drawing method更改为“ H bonds”,然后将“ Line width”更改为3,以便于查看。 同时将“Distance Cutoff”更改为4.2埃。 对于被认为是规则的氢键来说,这有点长,但这只是出于演示的目的,因为该轨迹不够长,无法形成任何真实的氢键。 现在,您的窗口应如下所示:
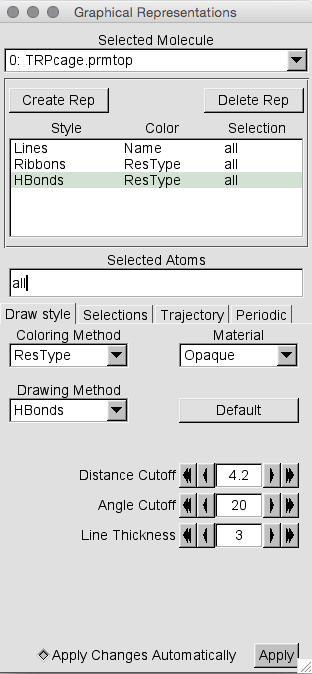
现在,所检测到的氢键将在所涉及的两个原子之间以虚线显示。 如果您浏览轨迹,您会发现它们是在各个地方形成的,然后被折断,然后重新形成。 这样,您可以直观地跟踪轨迹上氢键的存在。 请注意,如果这是一个显式的溶剂模拟,您可能希望将我们拥有的3种表示形式中的每一种的选定原子从“all”更改为“all not water”。 对于HBonds案尤其如此。 否则,您将被所有氢键水分子之间的虚线所淹没。
最后,我们将在肽链的两个末端之间添加一个键标签。 通过我们的模拟,这将允许我们监控链末端之间的距离。 转到“ VMD Main”窗口,然后选择Mouse-> Label-> Bonds。 这会将鼠标左键操作更改为bind选择。

然后转到“ OpenGL”窗口,然后在肽链的每一端单击一个原子。 您应该看到一条虚线,并显示它们之间的距离。 如果发现错误,可以使用Graphics->Labels 工具删除标签,以便再次创建标签。 注意:键,角和二面体标签选项实际上并不涉及键,角和二面体。 您添加的标签实际上不必在键合原子之间。 键表示任意两个原子之间的距离(以埃为单位),角度表示任意3个原子之间的角度(以度为单位),二面角表示任意4个原子之间的二面角(以度为单位)。
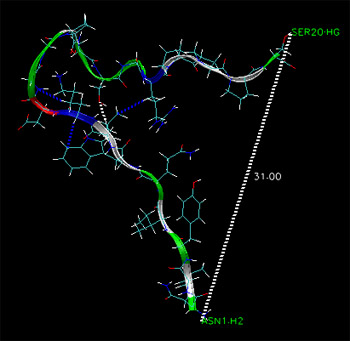
现在浏览轨迹,您应该看到值变化,并且TRPcage肽开始折叠。 我们在这里并没有真正了解该系统,但这只是为了向您展示VMD可以做的一些事情。 在更长的生产模拟中,我们可以学到更多并测量更多相关参数。
2.9 制作电影
最后,让我们学习如何创建具有轨迹的电影,以便我们可以在谈话中使用它。 注意,由于各种压缩编解码器的兼容性,这可能不适用于所有系统。 但是,请在您的系统上尝试一下,看看如何进行。 通过单击Extensions->Visualization->Movie Maker.。
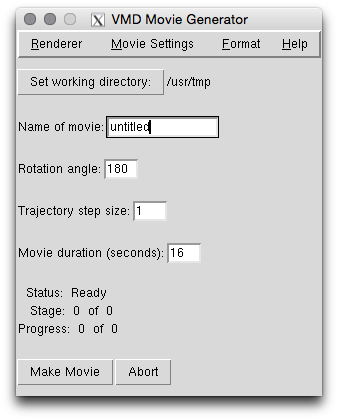
这里有很多可用的选项。我不会尝试涵盖所有选项,有关更多信息,请参见VMD网站。在此示例中,我将仅一步步创建一个简单的轨迹电影。首先选择合适的工作轨迹。此处的屏幕截图来自VMD的Unix版本。在Mac和Windows中,它看起来可能略有不同。
接下来输入电影的名称,例如TRPcage。接下来,我们需要根据所需的帧数选择影片的持续时间。电影将以每秒24帧的速度播放。 ( Format->Change Compression Settings for the frame rate)。因此,400帧将为我们的电影提供大约16秒的时间,这是合理的。如果我们希望它持续约8秒钟,我们可以将帧数减少到200,然后渲染其他所有帧。另外,我们可以将其设置为32秒,以获得800帧电影。电影长度是您应始终考虑的问题,因为它需要与您的讲话兼容。因此,在“Movie duration (seconds):”框中输入16。我们希望轨迹的每一帧,所以步长保持为1。

接下来,我们只想遍历轨迹,因此选择 Movie Settings->Trajectory。 然后在“格式”下选择“ MPEG-1”。 这将创建MPEG-1格式的视频文件。 如果需要,一定可以使用AVI格式播放。
然后点击"Make Movie”。 希望大约3分钟后,您应该在工作目录中找到一个电影文件。
这是TRPcage.mpg(2.9 mb)
本教程到此结束。 希望这为您提供了使用VMD查看和分析AMBER文件和轨迹所需的知识。 但是请记住,关于VMD可以做什么,我们甚至还没有scratched the surface。 如果您访问VMD网站,则会发现更多有关VMD中更高级功能的信息。
参考资料
