【2.5.3】amberTI介绍(thermodynamic integration)
在本教程中,将使用自由能计算来计算两个简单的配体苯和苯酚与T4-溶菌酶突变体L99A的相对结合自由能(relative binding free energy)。自由能将通过使用sander程序的热力学积分(thermodynamic integration)工具来计算。修改后的范德华方程(软核势,softcore potentials)用于确保平滑的自由能曲线。
本教程假定您熟悉Linux系统,Amber软件包的基本工作原理以及有关如何使用sander和pmemd运行MD模拟的知识。脚本的一些知识也将有用。
本教程适用于Amber10及更高版本的用户,因为TI的输入约定已在不同版本上进行了更改。从Amber14开始,现在还可以在pmemd中运行TI模拟以增强性能,但设置略有不同。较早的Amber版本不能用于此示例。关于TI早期版本的介绍,提供了有关计算pKa值或无溶剂化能量的教程。
本教程旨在为用户提供一个示例,说明有关如何原则上使用Amber软件套装执行自由能计算。它们不一定为特定应用领域提供参数或方法的最佳选择。
一、基本概念
1.1 生化背景 Biochemical Background
溶菌酶是一种14.4千道尔顿的酶(EC 3.2.1.17),它通过水解细胞壁的多糖成分来破坏细菌。 它富含多种分泌物,例如眼泪或多形核中性粒细胞的细胞质颗粒。 还发现它的浓度很高,例如 egg white。 作为最早结晶的一种酶,可以得到高分辨率的X射线结构(请参阅Blake等人,Nature 1965 761-763),它在数千个pdb数据库条目中得到引用。
在此示例中,我们将使用噬菌体T4溶菌酶的X射线结构(请参见此处的pdb条目),通过单点突变将适合小非极性分子结合的人工腔引入其中(请参见Eriksson等人,Nature 1992,371–373)。 已经确定了几种小分子对该酶的结合亲和力,使其成为研究蛋白质-配体结合的良好模型系统。 苯以-5.2 kcal / mol的ΔGBind结合在亲脂性腔中,而极性更大的苯酚则根本不结合。 假定结合后蛋白质没有发生主要构象变化,并且在不存在结合配体的情况下结合位点不含水分子。

T4溶菌酶L99A与红色的共结晶苯的X射线分辨率为1.8Å(vmd和povray生成
1.2 热力学循环与方法 Thermodynamic Cycle and Method

TI计算是通过参数λ耦合两个状态A和B来计算两个状态A和B之间的自由能差,该参数λ作为附加的非空间坐标。 这种λ形式主义允许将状态之间的自由能差计算为:
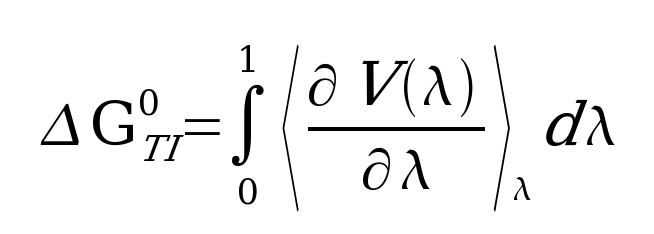
在该方程式中,V(λ)是与λ= 0时的V(A)和λ= 1时的V(B)相对应的λ耦合势函数。对积分的λ导数的平均值进行积分, 在给定的λ值下耦合势函数。 由于只能很少地通过分析来执行该积分,因此使用一种积分方案,其中在不同的离散λ点或“窗口”进行模拟,并通过数值计算积分的值。 TI计算的好处是,可以在固定的λ值下执行几个独立的MD模拟,从而在需要提高精度的情况下,可以进行有效的并行化并随后添加其他λ窗口。
由于热力学积分计算会计算两个化学组成不同的系统(例如,本例中为苯和酚分子)之间的自由能差,因此无法进行实验验证,因此必须使用类似于右图所示的热力学循环 将一系列TI计算的结果与物理观测值联系起来。 过程A和B代表两种不同的配体与蛋白质的结合,而过程C和D是从一个配体向另一种配体的转化,同时与蛋白质(C)结合或在水中简单地溶剂化(D)。 由于ΔGC-ΔGD=ΔGA-ΔGB,因此TI计算可用于计算相对结合自由能,使其成为药物设计或前导优化应用中的有用工具。
可以构造不同且更复杂的热力学循环来获得其他热力学量,例如。 溶剂化自由能或绝对结合自由能。 所有TI计算中的一般假设是,系统在转换过程中不会发生明显的构象变化,否则MD模拟很可能不会为收敛的结果采样足够的相空间。 如果需要计算构象变化产生的自由能,可以使用其他自由能技术。
使用sander和pmemd运行TI模拟的方式有所不同。 因此,以下内容可以分为两个步骤,但是鼓励读者阅读所有内容。 该教程的较旧版本仍然可用,因为sander的设置仍然相同,并且讨论也仍然非常相关。 较新材料的目的是提供一致的并排比较,并介绍如何在pmemd14及更高版本中运行TI。
1.3 一般说明
以下材料要求您熟悉Linux系统,Amber软件包的基本工作原理,并且具有如何使用sander和pmemd运行MD仿真的知识。一些脚本知识也将有用,因为大多数教程都希望通过现成的脚本来运行,以减轻通常繁琐且耗时的设置。这些脚本将利用所谓的HERE文档将输入传递给tleap和cpptraj,否则您将在交互式会话中手动输入这些输入。请从存储脚本的目录中运行所有脚本。
请注意,建议的模拟流程和详细的设置步骤不一定是最佳实践,也不是解决问题的唯一方法。所提供的脚本还很初级,不是很健壮,通常假定它们将运行而没有任何错误。但是,此处介绍的材料应该提供对基本概念的足够的见解,并让您开始在Amber中运行TI模拟。
1.4 教程概述
所有输入文件和脚本都可以在此处下载。 本教程位于配体子目录中,其中包含用于sander和pmemd的单独目录。 这些目录中的每一个都包含用于准备prmtop和起始坐标的设置,以及用于运行实际自由能模拟的free_energy。 后者中的脚本可能需要适应您的负载平衡软件或并行运行时环境。
基本的自由能方案将是一个三步方案,其中包括
- 苯的卸料步骤,decharging step of benzene
- 将(部分)排放的苯转化为(部分)排放的苯酚,transformation of (partially) discharged benzene to (partially) discharged phenol 3.以及最后对苯酚进行充电。 recharging of phenol.
第二步将被称为vdW + bonded步骤。
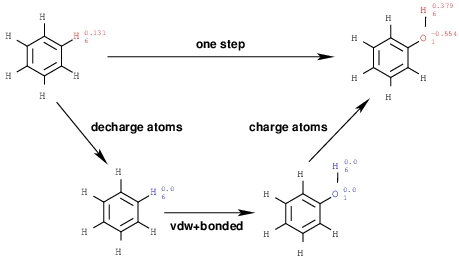
本教程中使用的三步循环是顶部一步突变的替代途径。 颜色中的原子标记了在模拟中进行转换的原子。 红色原子是带其原始部分电荷的带电原子,蓝色原子是带电的原子。 顶部的原子标签是部分电荷,底部的标签是索引号。 该协议必须在溶液中和在结合状态下均进行,以获得结合的ΔG。
该模拟系统将由不会进行变换且具有相同坐标集的原子组成。 这些将被称为共同原子(common atoms)。 所有要转换的原子都是TI区域的一部分,请参见下图。 在TI区域内,将存在以“单拓扑”方式直接从一种原子类型线性转换(LTA)到另一种原子并因此共享坐标(黑色原子)的原子。 消失或出现的原子以“软核”电势(SCA, “softcore” potentials )进行特殊处理,但原则上可以根据用户的需要将任何原子定义为软核原子。 这些原子不会彼此“看到”,因为它们之间永远不会发生相互作用(蓝色原子表示苯,绿色原子表示苯酚,数字标记是各自的原子指数)。
1.5 运行教程
- 如何使用pmemd设置TI模拟。 此功能自Amber14起可用。
- 如何使用sander设置TI模拟。 此功能自Amber10起可用。
- 由Thomas Steinbrecher(TSRI 2007)编写的原始教程可以在这里找到。 首先介绍如何设置参数和输入文件,然后继续进行分析。 此材料仍然非常相关,因为此后的输入几乎没有变化。 此外,本文讨论了新教程中未涵盖的各种角度。
- 迷你教程,演示如何针对侧链突变运行TI仿真。
二、使用Pmemd14及更高版本进行设置
所有输入文件和脚本都可以在此处下载。与本页上显示的材料相关的文件可以在 A9/pmemd/setup 中找到。鼓励您详细研究所有脚本以及输入和输出文件。
2.1 设置vdW + bonded转换步骤
脚本 1_leap.sh 将为此步骤创建prmtop和inpcrd文件。 Pmemd要求在prmtop文件中同时存在两种配体。因此,文件jump / bnz_phn.pdb 包含一组苯和苯酚的条目。配体将被放置在开始处,leap会将这些残基编号为1和2。这不是严格必需的,因为AMBER masks非常灵活,但是将配体放在明确定义的位置非常方便,特别是对于过程自动化。
实际上,我们在这里使用了一个技巧。 PDB文件中的碳原子都是LTA,这意味着它们必须共享相同的坐标。您有责任确保这一点。 Pmemd将在很小的公差范围内检查此情况,并告知您是否存在这种情况,然后终止。因此,在PDB中所做的就是将苯的配位复制到相应的苯酚条目中。 Leap将使用 jump/phen.lib 中的残基模板添加所有缺失的原子,从而创建有效的酚分子。使用您喜欢的可视化程序进行检查。
2.2 运行MD以提供合理的起始坐标
脚本 2_run_md.sh 将针对溶液中的复合物和两个配体进行最小化,加热和密度调节。 press.rst7 中的坐标将在所有后续步骤中使用。 输入文件中的协议乍看之下是TI协议。 我们在某种程度上任意设置λ= 0.5,但由于此变换的变化很小,因此相关性很小。 我们在这里主要要做的是创建一个密度调整的模拟框。
您可能需要调整此脚本,以根据系统上的需求并行运行作业。 缺省设置是在 apaches_prepare/和 complex_prepare /中都运行脚本run_all_md.sh,该脚本仅调用mpirun来运行pmemd。 您可能需要修改2_run_md.sh以调用上述子目录中的Submit.sh。 这是用于LSF作业计划程序的模板。 您可能必须更改此设置才能与其他调度程序并行运行作业。
2.3 从MD模拟中提取坐标
对于放电和充电转换(请参阅下面的步骤4),我们需要用两个拷贝的苯或苯酚替换两个配体。请运行 3_strip.sh 以执行此步骤。我们要用此步骤解决的问题是,在步骤#1中使用 solvateBox 时,leap将更改所有坐标,因此我们无法使用来自 jump/bnz_phn.pdb的坐标。因此,我们使用cpptraj来提取两个单独配体的正确坐标,并为系统其余部分提取坐标。
2.4 设置充放电步骤 Setting up the Decharging and Recharging steps
现在,我们已经从上面的步骤3获得了正确的坐标,并且可以从中组装出两个附加系统,并写出它们的prmtop和inpcrd文件。请运行脚本 4_leap.sh 以运行最后的设置步骤。该脚本在概念上与 1_leap.sh相同。我们将整个过程分为几个步骤的唯一原因是确保正确设置坐标。
运行TI模拟。
三、使用sander进行设置
旧版本,现已没在用
四、使用Pmemd或Sander运行TI模拟
所有输入文件和脚本都可以在此处下载。与本页上显示的材料相关的文件可以在 A9/pmemd/free_energy 和 A9/sander/free_energy 中找到。鼓励您详细研究所有脚本以及输入和输出文件。
4.1 准备输入文件并运行TI模拟
如下所述,如何使用pmemd或sander运行TI模拟存在一些差异。但是,让我们首先转到常见的准备步骤。
脚本setup.sh将创建所有输入文件,并链接到prmtop并从设置步骤开始inpcrd。对于溶液中的每个系统,络合物和配体,将创建一个目录。在这三个步骤的每个三个目录中,将创建:decharge /,recharge /,vdw_bonded /。在这三个目录的每个目录中,将为每个λ值创建目录。输入文件将从模板创建,唯一的区别是是否使用了clambda,蒙版和其他软核原子(仅在vdw + bonded步骤中)的值。
脚本Submit_ligands.sh和Submit_complex.sh可用于将所有作业提交到作业计划系统。您可能必须调整这些脚本。这些只是用于LSF作业计划程序的模板。这些作业可能会运行几个小时,具体取决于您的硬件,负载等。
桑德(Sander)需要以一种特殊的模式运行:multisander,它可以同时运行多个模拟系统。对于TI计算,这意味着两份副本,每个状态一份。这也意味着核心数必须是2的倍数。此外,最小化只能在两个核心上运行,而MD模拟可以根据需要使用任意数量。
multisander的命令行指定组数(-ng)和组文件。然后,该组文件在每个组(即两个)的一行中都包含命令行参数,就像用于sander的常规参数一样。 vdw + bonded步骤的示例从初始状态文件读取组1的prmtop和inpcrd文件,从最终状态的相应文件读取组2的prmtop和inpcrd文件。
-O -i ti0.in -p ti0.parm7 -c ti0.rst7 -inf ti0_0.info -o ti0_0.out -r ti0_0.rst7 -e ti0_0.en -x ti0_0.nc
-O -i ti1.in -p ti1.parm7 -c ti1.rst7 -inf ti1_1.info -o ti1_1.out -r ti1_1.rst7 -e ti1_1.en -x ti1_1.nc
analyse.sh脚本可用于对完成的MD运行进行快速分析,请参阅下一节。
4.2 Pmemd和Sander之间的共性和差异
这两个程序之间的共同点是通用MD协议和大多数自由能设置。真正的区别仅源于以下事实:pmemd仅需要一个输入文件和一个包含两个TI区域的prmtop文件,而sander分别为每个TI区域读取两个输入文件和两个prmtop文件。这两个程序都可能需要一个软核屏蔽,以及一个可选的电荷屏蔽。
桑德(Sander)没有TI遮罩,因为它同时复制了非转化和非软核原子。仅软核原子需要掩膜。不必要的重复是pmemd中重新实现的原因之一,它可以消除这种情况,但还要求用户明确定义TI区域(每个区域两个掩模),其中仅包含转换的原子。另一个原因是sander不允许采样最终状态,即λ= 0.0或1.0。有关详情,请参见 DOI:10.1021 /ct400340s。
4.3 输入文件的详细信息
样本输入文件如下所示。
&cntrl
imin = 0, nstlim = 100000, irest = 1, ntx = 5, dt = 0.002,
ntt = 3, temp0 = 300.0, gamma_ln = 2.0, ig = -1,
ntc = 2, ntf = 1,
ntb = 2,
ntp = 1, pres0 = 1.0, taup = 2.0,
ioutfm = 1, iwrap = 1,
ntwe = 1000, ntwx = 10000, ntpr = 10000, ntwr = 20000,
icfe = 1, clambda = 0.1, scalpha = 0.5, scbeta = 12.0,
ifmbar = 1, bar_intervall = 1000, bar_l_min = 0.0, bar_l_max = 1.0,
bar_l_incr = 0.1,
<sander vs pmemd specific parameters, see below>
/
以上是与sander和pmemd相同的部分。 请查阅手册以获取有关模拟协议的详细信息,尤其是空行上方的控制参数。 另外,请查看通过本教程生成的示例输入。
要打开TI ifce必须将其设置为1,clambda是当前的λ,scalpha和scbeta是软核参数。 如果mbar = 1,则打开(M)BAR后分析的输出(另一种获取自由能的方法,请参见DOI),这两条线上的其他参数控制该输出的频率,并应针对λ值执行此操作。 另一个参数是ifsc,它控制是否使用软核参数。 对于线性的去充电和再充电转换,这将是ifsc = 0,因为在此步骤中没有原子出现或消失。
4.4 定义TI,软核和电荷掩膜 Defining the TI, softcore and charge masks
Pmemd仅读取一个输入文件。 这里是vdw + bonded步骤的蒙版。
timask1 = ':1', timask2 = ':2',
scmask1=':1@H6', scmask2=':2@O1,H6', crgmask=':1@H6 | :2@O1,H6'
Pmemd需要明确定义TI区域,并且每种状态下的软核原子都有两个掩码。 电荷掩膜是每个单独的配体电荷掩膜的并集。
桑德(Sander)没有TI地区的概念(请参见上面的讨论)。 第一个过程(初始状态)的输入为
scmask=':1@H6', crgmask=':1@H6'
第二个过程(最终状态)的输入为
scmask=':1@O1,H6', crgmask=':1@O1,H6'
五、分析结果
analyse.sh脚本可用于分析TI梯度(dV / dl)并通过积分计算ΔG。 MD运行必须已经完成。 该脚本运行Python 2.x代码,并且还需要安装Numpy。
由于Sander无法采样端点,因此脚本将进行6阶多项式拟合,并根据该拟合估计λ= 0和1的梯度。 另外,脚本可以根据两个最近的点进行线性外推(请参见getdvdl.py中的源代码)。 所有 dV/dl 梯度将根据λ写入相应目录中的文件 dvdl.dat。
您的输出可能看起来像这样
ligands/decharge: -6.44585036422
ligands/vdw_bonded: 1.29181321994
ligands/recharge: -33.8336526019
--------------------------------
dG sum for ligands = -38.98768974618
complex/decharge: -7.14129064364
complex/vdw_bonded: 4.74879707081
complex/recharge: -34.3999259026
--------------------------------
dG sum for complex = -36.79241947543
结合的最终ΔG为ΔGcomplex-ΔGligands,在这种情况下约为+ 2.2kcal / mol。 但是请注意,由于本教程中使用的采样有限,并且可能是由于选择了lambda时间表,因此您获得的结果可能会有所不同。 在如此短的模拟时间内出现1甚至更大的kcal / mol的偏差不应是意料之外的。 然而,该结果表明,如实验所预期的,苯比苯酚与T4溶菌酶的结合更强。
Thomas Steinbrecher的原始教程对结果进行了更深入的分析和讨论。 但是请记住,在实际工作中,TI模拟将需要运行更长的时间,并且详细的协议可能需要进行调整。
六、最后讨论
6.1 一步骤和多步骤协议
配体教程与原始教程一样,使用三步协议(放电,vdw +键合,充电)协议。这是一种非常通用的协议,当两种状态都具有虚拟原子(在转换中出现或消失的原子)时,可以使用该协议。 pmemd和sander的现代版本都允许Lennard-Jones相互作用和静电相互作用具有软核势。实际上,默认情况下,两个软核潜力都处于打开状态。这意味着任何转换原则上都可以一步完成。这似乎对绝对转换效果很好(例如DOI:10.1063/1.2799191)。从技术上讲,您可以将输入文件用于vdw + bonded转换,而只需删除crgmask。
但是,对于相对转换,本教程的作者发现单步协议在某些情况下可能会产生错误的结果。因此,在使用此协议时应格外小心。最终,所产生的自由能必须与所选择的路径无关。
如果选择的路径导致闭合循环,当然也可以使用编号不同的步骤协议(路径)。例如,将静电转化与vdW +键合的静电转化分离是相对容易的。当出现或消失的原子仅以一种状态存在时,此协议有效。其他协议可能涉及不同的原子映射或软核原子的不同选择(但最小数目必须是出现/消失的原子)。
6.2 相对和绝对自由能
本教程中介绍的炼金术自由能(alchemical free energy)计算方法是相对自由能计算。这为我们提供了苯与苯酚在T4溶菌酶中的相对结合亲和力,因此该方法可用于排名。所谓的绝对自由能将直接测量结合自由能。从技术上讲,您需要通过将所有原子定义为软核原子来完全“破坏”溶液和复合物中的配体。由于最终状态是不存在的分子,因此pmemd prmtop将仅具有一个配体(并且TI遮罩在一个末端状态设置为空字符串),而在sander中,一个prmtop将具有配体,而另一个则没有。一步协议似乎可以正常工作,请参阅[DOI:10.1063 / 1.2799191](alchemical free energy)。
但是,绝对自由能模拟的收敛速度可能比相对模拟慢,并且统计误差通常更大。另一个复杂因素是,结合的最终状态定义了一个不存在的分子,由于不存在vdW(可能还有静电)相互作用,该分子在模拟系统的整个空间内自由漂移。这违反了微观可逆性。为了解决该问题,引入了附加的约束以将结合的配体保持在适当的位置。这意味着需要对系统进行额外的工作,并且还要确定相关的自由能。参见例如 DOI:10.1021 / ct3008099
6.2 单拓扑和双拓扑
术语“单”和“双”拓扑是指在拓扑中如何表示系统的转换区域的方式。实际上,sander和pmemd都是混合体,用户可以在两个极端之间无缝切换。软核原子以双重拓扑方式处理,而其他原子则视为单一拓扑。单一拓扑意味着只有一组坐标。软核原子始终两次表示为“唯一”原子,这意味着坐标对于每个状态都是唯一的。这就是为什么必须通过使单个拓扑原子在拓扑和坐标文件中具有相同顺序来映射它们的原因。这里的映射意味着等价原子成对配对,例如苯中的C1到苯酚中的C1等,但请注意原子标记是任意的。
在实践中,限制软核原子的数量(但最少要包括所有出现和消失的原子)并假定大多数原子可以直接映射是有用的。典型的应用是对通常具有常见刚性骨架的类药物分子进行前导优化。然后可以将此支架视为单个拓扑,因此两个状态都将具有单个坐标集。在低相似性的情况下,该方法将需要更高程度的双重拓扑。极端的情况是采用“全”双拓扑结构,但随之而来的是漂移配体的问题,与绝对自由能讨论的一样。实际上,可以将非共价键合分子的双重拓扑模拟视为两个同时的绝对模拟(但只能获得相对自由能)。
需要强调的另一点是,炼金术自由能模拟旨在通过一个封闭的热力学循环进行。从一个循环中的单腿获得的自由能相对没有意义。您可以通过将不同组的原子定义为软核来进行尝试。由于涉及软核原子的力场项不会计入TI梯度,因此您每次更改软核原子时都会计算出不同的自由能。最终的ΔG应始终约为相同(前提是可以对系统进行充分采样)。假定对自由能的某些贡献将在一个周期内抵消(或小到无所谓)。
6.3 自动化
您可能现在已经知道,仅一次TI模拟的设置可能非常繁琐,并且很容易出错。 本教程提供的脚本表明,某些脚本可以非常有助于加快流程速度并避免潜在的陷阱。 但是,有一些代码旨在使相对的炼金术计算的设置变得更加容易,并在可能的情况下使过程自动化。 其中一个代码是Free Energy Workflow Tool(FEW),它是AmberTools的一部分,有关介绍,请参见教程24。 另一个代码是独立开发的FESetup( DOI:10.1021 / acs.jcim.5b00368)。由本教程的作者撰写。 FESetup支持各种多步骤协议,还支持显式虚拟原子。 网络资源 一个推荐且有用的资源是Alchemistry.org,它提供了许多有关自由能模拟的背景信息,并且还提供了一些教程来实现比此处描述的更为复杂的任务。
参考资料
