【7.1】药物发现中炼金术自由能方法的发展
本手稿的目的是简洁地追溯炼金自由能法(AFEM,alchemical free energy methods)从纯理论构造到现在广泛用于生物技术和制药行业的方法的发展中的关键技术步骤。 更具体地说,我们关注的是相对结合自由能(RBFE,relative binding free energy)计算,这种计算更多地应用于计算机辅助药物设计(CADD)活动,而不是计算量更大的绝对结合自由能(ABFE, absolute binding free energy)计算。 我们在时间轴的开发过程中并未穷举,而是尝试编织一个有关理论思想如何最终转化为当代自由能源能力的故事。 这种讲故事的方法必然会限制我们引用有关AFEM的所有工作,对此我们深表歉意。 但是,对于那些对广泛描述该领域中所有工作感兴趣的人,他们将针对许多现有的出色评论。
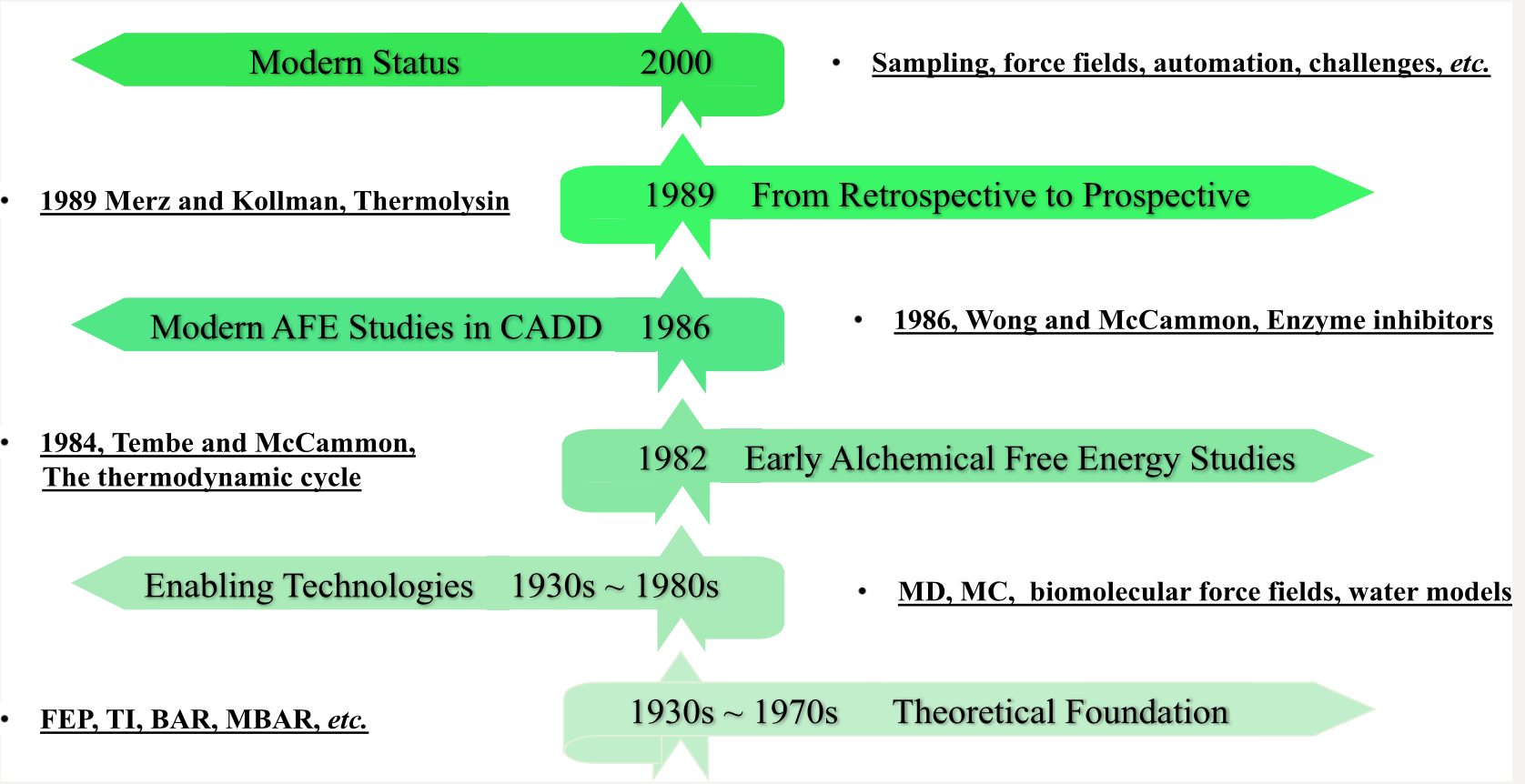
一、 理论基础 THEORETICAL FOUNDATIONS
尽管自由能摄动(FEP,free energy perturbation)方法仅在1980年代引起计算机辅助药物设计(CADD)领域的注意,但其理论基础是30年前奠定的。 在对非极性气体的性质进行理论探索期间,Zwanzig推导出了FEP的主方程,尽管采用热力学扰动理论的研究可以追溯到1930年代的Peierls。FEP方程的内在美(参见等式1
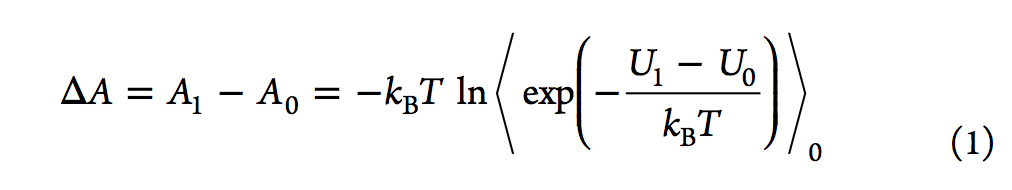
在于以下事实:通过对状态0的势能引导的配置进行采样,然后计算采样状态下两个状态的势能,可以计算两个整体状态(状态0和状态1)之间的自由能差配置。在等式1中,⟨⟩0 代表在状态0下的玻尔兹曼平均值,T是温度,kB是玻尔兹曼常数,U1和U0分别是状态1和状态0的势能。在这里,我们使用亥姆霍兹(Helmholtz)自由能差(ΔA)代替吉布斯自由能差(ΔG),但在ΔPV值通常较小的凝聚相模拟中,两者的差异可忽略不计。
从表面上看,使用该主方程似乎很简单,但FEP计算的收敛性并不容易实现,尤其是对于两个端态集合的平衡分布几乎没有重叠的系统。然而,柯克伍德(Kirkwood)在1935年[13]描述的使用耦合参数的想法极大地有利于FEP计算。简要地,经由耦合参数λ引入一系列中间状态。随着λ从0增大到1,中间状态的势能(Uλ)从U0变为U1。然后计算相邻状态之间的ΔAs,所有ΔAs的总和得出总ΔA。用于将中间状态定义为λ函数的电势函数的典型形式,是参考状态U0的电势与U1的电势的线性组合(请参见等式2)。
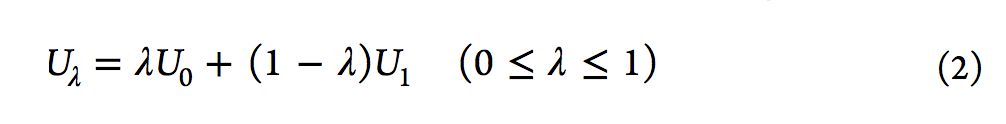
有效估计自由能差的另一种方法是Bennett接受率(BAR,Bennett acceptance ratio),该方法由Bennett在1976年提出。通过类似于FEP方程,他推导出了等式3,其中引入了加权函数(w)。
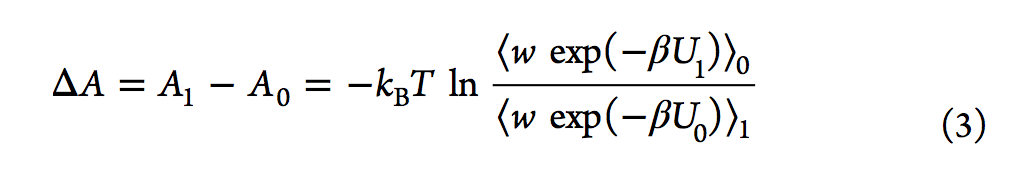
通过最小化自由能差的期望平方误差,贝内(Bennett)特获得了导致方程4的最佳加权函数,可以通过对ΔA进行迭代试验来求解。
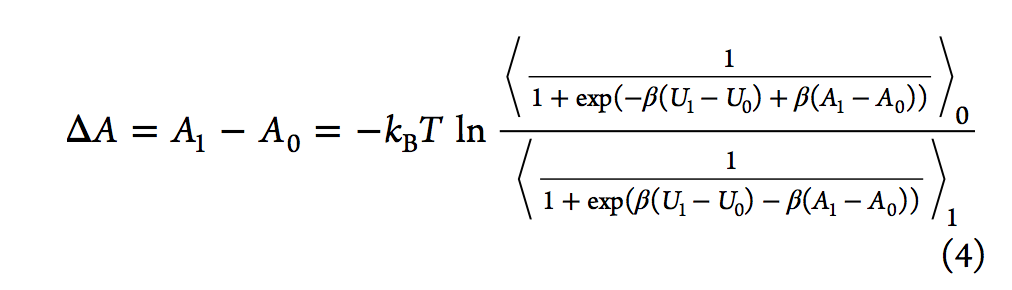
与Zwanzig的方程式不同,BAR分析需要在两种状态下进行采样配置。 2008年,报告了多状态贝内特接受率(MBAR,multistate Bennett acceptance ratio)方法,该方法通过结合来自多个状态而不是仅来自两个状态的模拟数据来估算自由能。另一种常规方法是基于柯克伍德的热力学积分(TI)。 它通过对状态λ相对于λ的玻尔兹曼平均势能导数进行积分来计算ΔA(请参见等式5).

可以在文献中找到可用的自由能方法范围的更详细的推导和解释。
二、启用技术 ENABLING TECHNOLOGIES
为了执行炼金自由能(AFE, alchemical free energy )计算,基于理论公式,需要生成代表性结构并评估相应的势能。因此,仿真技术和力场的发展成为实施AFEM的关键因素。
- 一种主要的采样技术是蒙特卡洛(MC, Monte Carlo)仿真。简而言之,MC模拟从给定的配置开始,并以最终采样的配置恢复实际整体的真实分布的方式接受或拒绝下一个建议的配置。 MC方法可以追溯到Enrico Fermi,尽管最早发表的工作示例是由Ulam和Metropolis进行的。第一篇将MC方法应用于分子研究的论文发表于1955年,并且蛋白质的MC模拟是1970s报道。
- 另一种主要的采样技术是分子动力学(MD,molecular dynamics)模拟。 MD仿真涉及沿时间轴的采样配置( sampling configurations)。 1964年,拉赫曼(Rahman)发表了关于液态氩的具有里程碑意义的MD模拟,他在那儿使用Lennard-Jones势来描述氩原子。随着适用于生物分子的力场的发展, McCammon,Gelin和Karplus于1977年报道了对蛋白质的首次MD模拟,该模拟基于Levitt和Warshel在Lifson实验室开发的模拟程序。Berendsen同年,开发了“ SHAKE”算法,该算法限制了高频重原子氢键,这一贡献使得将MD模拟中使用的时间步长增加了2倍,从而可以进行更广泛的采样可用的计算机资源。另外,算法的实施可以保持恒定的温度和恒定的压力,例如,Berendsen和同事在1984年的耦合方法,使得MD仿真可以对物理上实际的配置进行采样。
生物分子力场的发展是基于Lifson实验室在1960年代后期的开创性工作[26,31,32],关于势能函数的形式参见式6。前三个项分别表示键,角度和扭转对势能的贡献,而后两个项分别表示范德华力的非键合和成对静电相互作用。在等式6中,Kb,Kθ和Kφ是键,角度和扭力常数; b,θ和φ表示键合长度,角度和扭转角; b0和θ0是参考键的长度和角度。 n是周期性,δ是相位。 ε是井深(well-depth),r0是伦纳德·琼斯极小值的位置;最后,qi和qj分别是原子i和j的原子电荷。在1980年代,还根据Lifson的力场模型开发了其他早期的力场和程序,包括Karplus等人的CHARMM33,Kollman等人的AMBER,Jorgensen等人的OPLS36和Berendsen和van Gunsteren的GROMOS37。等。简而言之,对键,角,二面体,电荷和Lennard-Jones(LJ)参数进行了优化,以重现量子力学(QM)或实验性质。在过去的几年中,许多努力集中在改进力场模型上,包括创建可极化的力场。可以在许多评论中找到当前力场最新技术的概述以及详细的历史观点。
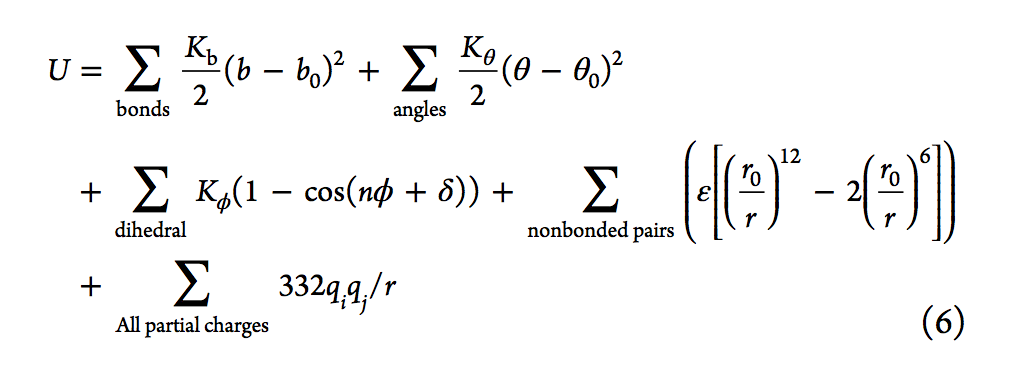
用于AFE计算的另一项重要的使能技术(enabling technology)是开发强大而准确的水模型。药物的绝对结合自由能和相对结合自由能都受溶剂影响。因此,对水和小分子的溶剂化进行精确建模至关重要。可以使用隐式水模型(例如Poisson-Boltzmann 或Generalized Born 模型)来模拟溶剂对药物结合的影响,但是许多水受体或水-药相互作用是特定的,因此需要使用显式水分子以获得最高质量的结果。例如,活性位点水的置换在自由能预测中可以发挥巨大作用。
根据使用了多少个额外的带电虚拟原子(没有vdW势项),水模型可以分为三个位点,四站点,五站点和六站点模型。最早的水模型是1933年由Bernal和Fowler描述的BF水模型,由Ben-Naim和Stillinger于1972年描述的BNS水模型,以及由Rahman和Stillinger于1974年描述的ST2水模型。约根森一直是开发实用水处理技术的先驱。以及用于分子模拟的精确水模型。他开发了TIP模型(可转移的分子间电势)来创建用于水,酒精和乙醚模拟的溶剂模型。通过MC模拟获得了气相二聚体和纯液体的合理结构和含能结果。同样在1981年,Berendsen及其同事设计了SPC(单点进水,simple point charge)水模型。拟合参数以重现300 K时的实验相互作用能和液体压力。Jorgensen继续开发和修改TIP水模型,创建了TIPS2,TIP3P和TIP4P水模型。这些水模型都是与一系列实验结构和热力学特性进行了比较,这些特性为将来对水模型的评估奠定了基础。 Berendsen通过包括自我能量校正的方法来发展他的SPC模型,从而创建了SPC / E水模型。迄今为止,TIP3P和SPC / E水模型是使用最广泛的模型,但仍在开发更精确的水模型一个活跃的研究领域。对于那些对水模型的发展和演化感兴趣的人,可以进行几篇总结水模型发展状况的评论。
三、早期的炼金自由能法研究 EARLY ALCHEMICAL FREE ENERGY STUDIES
有了所有使能技术(enabling techniques),AFEM的应用就会切实开始。 Postma,Berendsen和Haak在1982年[62]报道了FEP对水中空腔形成的计算。这项工作几年后,Jorgensen和Ravimohan于1985年对代表性分子进行了FEP计算。他们计算了稀溶液中甲醇和乙烷的相对水合自由能,其结果令人兴奋,与实验吻合。这提示了CADD中FEP计算的潜力,因为相对溶剂化自由能在确定一个共同受体位点上两个配体的相对结合自由能中起主要作用。当然,对水,乙烷和甲醇的潜在功能的优化对于这项工作的成功至关重要。除了将FEP应用于相对水合自由能计算外,Jorgensen及其同事还利用FEP为SN1反应((CH3)3CCl的解离)建立了潜在平均力(PMF)分布图,打开了通往研究溶剂化对反应过程的影响。乔根森(Jorgensen)的工作启发了科尔曼及其同事从AMBER程序套件中将FEP实施到其MD仿真程序中,并且他们运用FEP / MD方法研究了各种系统,包括有机分子和蛋白质抑制剂系统。作为最早的应用之一,他们在水中干扰了甲醇对乙烷,氢鎓对铵,甘氨酸对丙氨酸,丙氨酸对苯丙氨酸的干扰,其计算结果与现有的实验数据基本吻合。TI方法最初由柯克伍德提出,包括Mezei,Berendsen和其他几个小组的作者也将应用于各种系统。

这些早期研究显示了 AFEM 在不同系统上的潜力。但是,使AFEM广泛应用于CADD的主要贡献是实现了热力学循环的吉普斯或亥姆霍兹自由能等状态函数的幂,从而简化了相对结合自由能(RBFE)计算的计算。图1所示的上部和下部过程分别表示配体L1和配体L2与受体位点R的绝对结合自由能(ABFE)。 ABFE计算具有挑战性,并且计算量很大,因此需要简化。在配体优化工作中,通常对支架进行简单的修饰(例如-H被-CH3取代),因此最后重要的是这些化学扰动下的相对自由能变化。通过连接图1的上部和下部过程,我们发现ΔΔA=ΔA1-ΔA2可以通过两个垂直过程表示的炼金术转化获得ΔΔA=ΔA3–A4。因此,两个配体的RBFE可以通过以下两个炼金过程的AFE计算获得:R + L1→R + L2和RL1→RL2。
以这种方式转换问题会将两个计算量大的计算转换为两个计算量大的AFE计算。 Tembe和McCammon在1984年发表的题为“配体-受体相互作用”的论文中,首先在RBFE计算的背景下提到了热力学循环的概念。在本文中,他们设计了一个模型系统,并将热力学循环与FEP计算结合起来进行计算。两个“配体”原子的ΔΔA。他们的结果与通过伞式采样法直接计算ΔA1和ΔA2自由能而获得的ΔΔA非常吻合。 McCammon和他的同事还应用了“热力学循环扰动法”来计算Cl-和Br-离子的相对水合自由能,与实验结果吻合良好。更重要的是,该方法已应用于计算有机受体SC24的Cl-和Br-离子的RBFE,并且与实验结果吻合良好。这项工作由Lybrand,McCammon和Wipff于1986年出版,首次证明了AFEM通过热力学循环在客座系统(host−guest systems)上的适用性。值得注意的是,约根森(Jorgensen)于1985年所做的有关甲醇和乙烷的相对水合自由能的研究也利用了热力学循环的概念。通过将配体表示为刚性的,即不具有分子内自由度,通过在水溶液中将一个配体彼此干扰而获得相对的溶剂化自由能,而在气相中则没有扰动,因为其设计值为零。除了使这些计算更容易处理之外,热力学循环概念还提供了误差消除功能,从而提高了计算出的自由能的质量。
使用RBFE计算的研究暗示了CADD中AFEM的前景光明。除了RBFE研究之外,还进行了ABFE计算。在他的工作中,科尔曼通过使用FEP消除了一个核酸碱基,在真空中计算了两个核酸碱基的ABFE。结合两个碱基的水合自由能,他能够通过热力学循环获得水中两个碱基的ABFE。约根森还利用热动力循环推导了双重an灭方法(DAM,double annihilation method )。简而言之,对RL1→R和L1→0这两个炼金过程进行FEP计算以获得甲烷二聚体的ABFE。基于这种方法,已报道了来自多个组的多项研究.84− 90一个重要的例子是由Gilson等人[2],Roux等人[91],Hermans等人[87]和Boresch and Karplus等人[89]提出并提出的双重去耦方法(DDM)绕过了端点采样DAM中使用的RL1→R过程中存在一个问题,方法是引入一种中间状态,其中使用约束条件将解耦的配体约束在适当的位置。尽管进行了许多研究ABFE的研究,但RBFE计算在CADD中更加常规地执行,因为它通常更准确且计算量较小。因此,如摘要所述,我们将重点研究用于计算CADD中相对结合自由能的炼金术自由能方法的发展。
四、CADD中的现代AFE研究 MODERN AFE STUDIES IN CADD
McCammon及其同事在卤离子与SC24受体结合方面的工作通过热力学循环证明了FEP在宿主系统(host−guest systems)中的适用性。在同一年,Wong和McCammon将他们的研究扩展到了酶抑制剂系统。 JACS于1986年1月收到了他们的著作《酶和抑制剂的动力学与设计》,这是CADD中的第一个现代AFE研究。在该研究中,Wong和McCammon计算了对氟苯甲酰胺和苯甲与制胰蛋白酶的RBFE,benzamidine对天然和突变胰蛋白酶的RBFE,使用GROMOS程序。尽管由于采用了简短的模拟方法而具有较大的不确定性,但所计算出的ΔΔA值却与实验相吻合,证明了“热力学循环扰动法”在CADD中的适用性。 1986年下半年,在AMBER程序套件中实施FEP方法后,Kollman及其同事研究了一对结合至嗜热菌素的抑制剂(即膦酰胺和膦酸酯),计算得出的ΔΔA值为4.21±0.54 kcal /摩尔,与4.10 kcal / mol的实验值非常吻合。对于每个扰动,还进行了逆变换以探索计算结果的收敛性。较小的不确定性(ΔΔA的13%)和与实验的紧密一致性令人鼓舞。重要的是,在来自McCammon和Kollman组的两项研究中,抑制剂的相对溶剂化自由能对计算出的ΔΔA做出了重要贡献,这进一步说明了准确描述有机分子与水之间相互作用的重要性。随后,Kollman及其同事将研究扩展到除了配体结合之外,还进行了位点特异性诱变对酶催化的影响。他们计算了天然和突变枯草杆菌蛋白酶以及三枯草杆菌蛋白酶对三肽底物的相对活化自由能,相对结合自由能。在实验结果被披露之前,他们正确地预测了结合自由能的微小差异以及激活自由能的实质差异。经过1980年代的初步研究后,CADD的AFE研究在1990年代扩大了,报道了关于根霉胃蛋白酶,胰凝乳蛋白酶,弹性蛋白酶,HN-1蛋白酶,碳酸酐酶II,二氢叶酸还原酶和T4溶菌酶的研究。尽管有MC模拟在宿主系统的早期FEP研究和相对溶剂化自由能的计算中取得了成功,由于围绕采样效率的问题,其在蛋白质模拟中的使用受到限制。在一个蛋白质区域中,小幅度试探骨干角可能会导致远端部位大幅度移动。因此,总体接受率将很小。 Jorgensen于1997年对CADD进行了前两次现代MC / FEP研究。与实验取得了很好的一致性,但两项研究均使蛋白质骨架保持固定。 MC / FEP的突破是在2002年实现的,当时Ulmschneider和Jorgensen在MCPRO程序中实现了带有柔性键和二面角的协调旋转算法,用于MCPRO程序在蛋白质配体系统上中的MC模拟。。
五、从追溯到预期 FROM RETROSPECTIVE TO PROSPECTIVE
即使McCammon,Kollman,Jorgensen等小组的早期工作显示了AFEM在CADD中的潜力,但仅报道了回顾性研究,其中事先知道了抑制剂的实验结合自由能。在CADD应用中,实验结果尚不清楚,因此,为了使这些方法在CADD中找到合适的位置,必须证明它可以做出准确的前瞻性预测。 1989年,Merz和Kollman首次报道了蛋白质-药物样配体系统的前瞻性研究,首次预测了新抑制剂的结合自由能,随后通过实验对其进行了验证。他们研究的蛋白质是嗜热菌蛋白酶,抑制剂具有碳苯并氧杂-Glyp(X)-Leu-Leu的一般结构,其中X = NH,O和CH2。在较早的工作中已经研究了NH和O化合物[93],但是它们的实验结合自由能是事先已知的。对于新的抑制剂,即CH2化合物,Merz和Kollman正确地预测它具有与NH化合物相似的结合自由能(0.0 kcal / mol的计算值与-0.1 kcal / mol的实验ΔΔACH2→NH),这令人惊讶地大于O化合物的结合自由能。值得一提的是,他们还探讨了电荷选择对药物分子和关键扭转参数的影响,对计算出的ΔΔA值产生了影响。扭转参数和电荷组的优化仍然是提高CADD中AFEM精确度的一项持续努力。两年后,1991年,Kollman及其同事在CADD中进行了第二项前瞻性FEP研究。他们研究了与肽抑制剂结合的肽抑制剂。 HIV-1蛋白酶正在进行ΔΔA值的实验确定。抑制剂JG365的S和R非对映异构体的预测ΔΔA与实验一致。
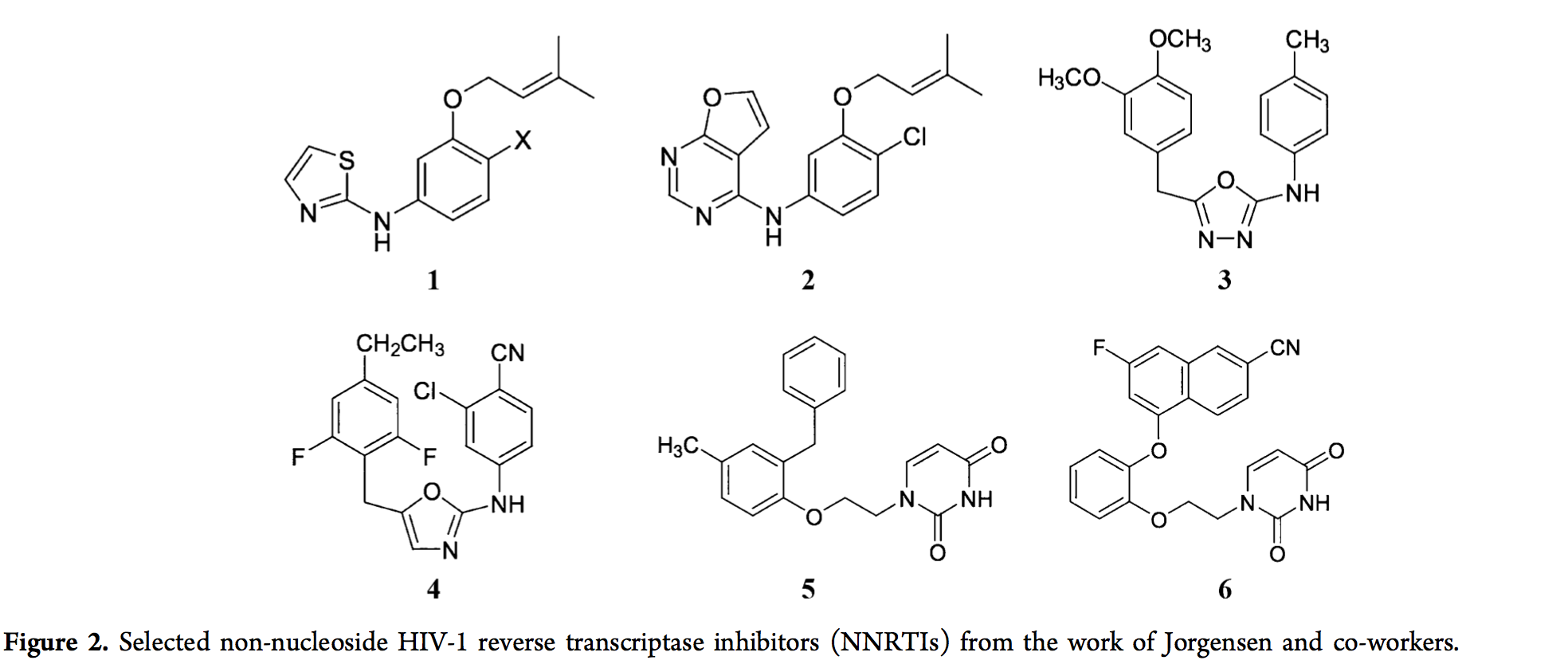
迄今为止,已经报道了广泛的前瞻性CADD研究。当前观点的目的不是详尽地列举所有现有研究。尽管如此,我们将重点介绍一些示例。从21世纪初期开始,Jorgensen及其同事开始利用FEP指导非核苷HIV-1逆转录酶抑制剂(NNRTIs)的设计,并且他们还通过实验测量了其活性(EC50)和细胞毒性(CC50),验证来自FEP计算的预测。2006年,从lead化合物1(Het-NH-PhX-U)开始,参见图2,MC / FEP计算(在MCPRO程序中实现)指导了几种NNRTI的设计。通过排列1中的Het和X基序,具有与两个FDA批准的NNRTIs类似的活性和细胞毒性,即依非韦伦和依曲韦林。 新型NNRTIs之一是化合物2,其EC50为5 nM,CC50为17微米与具有10μMEC50的初始lead化合物1(X = H)相比,其效力得到了极大的提高。从2008年开始,其中包括重要的突变来检测NNRTI的活性,例如Y181C。发现最好的先前化合物对这些病毒突变具有低活性或无活性。但是,Jorgensen及其同事能够使用MC / FEP计算来指导他们在新的NNRTIs [106,115]上的发现,从2007年发现的具有U-Het-NH-PhX结构基序的新导线(化合物3)开始。 前导化合物3是通过虚拟筛选获得的假阳性化合物,但对U,Het和X基团的干扰会导致产生真正的阳性化合物4,该化合物对野生型和Y181C突变体的EC50为1.3 nM和6.9 nM转录酶。 2009年,MC / FEP计算从筛选出的能够突出Y181C和野生型活性的先导化合物(化合物5)开始,引导他们找到了对野生型和重要突变体均具有活性的有前途的化合物,即Y181C和K103N / Y181C,通过取代模式,连接子区域和杂环中的扰动引起。一个实例是化合物6,其对于野生型,Y181C突变体和K103N / Y181C突变体转录酶的EC50为1.1 nM,8.0 nM和6.0 nM,并且其CC50大于100μM。再次获得了化合物5的显着改善,其野生型EC50为4.8μM,两个突变均无活性。经过多年的努力,基于MC / FEP的计算已帮助Jorgensen及其同事将针对野生型和重要突变体转录酶的针对NNRTI的EC50从μM水平提高至nM水平。在小鼠上的临床前试验表明,所设计的NNRTI的功效。
最近,Lovering等人使用相同的方法。能够预测脾酪氨酸激酶的17种潜在化合物中活性最高的lead化合物,进一步优化该lead化合物可产生nM细胞活性。最后一个例子是,Janssen Pharmaceuticals在2017年采用FEP +程序研究β-分泌酶1(BACE1)系统。通过FEP评估了抑制剂的支架和P3口袋取代基上的修饰,所得抑制剂表现出nM活性。 FEP结果与P3口袋中取代基修饰实验之间的平均无符号误差(MUE)为0.35±0.13 kcal / mol。
尽管列出了成功的示例,但AFEM仍需要解决许多问题,才能使例行可靠地使CADD受益。即使在以前的研究中,也报告了AFE计算中的缺陷。例如,在Lovering等人的研究中,尽管大多数情况下相对自由能变化的迹象是正确的,但FEP计算出的ΔΔA值通常比实验ΔΔA大得多。此外,在Janssen Pharmaceuticals报告的工作中,很难实现支架修饰的融合。在下一部分中,将进一步讨论如何进行改进以及提高AFEM在CADD中的作用和准确性所需要的改进。
六、现代状态 MODERN STATUS
自从AFEM在CADD中首次应用以来已经有34年了。从其理论基础出发,实现AFEM的程序已经发展到不仅支持学术研究而且还支持药物设计运动。 在过去的20年中,随着计算机功能的增强和对计算研究的兴趣,为改进CADD的AFEM付出了巨大的努力。 我们无法总结所有贡献,但将重点介绍一些显着的发展,包括采样技术的改进,力场的改进和自动化。 除了所做的改进之外,我们还讨论了一些仍需要解决的问题,以提高AFEM的效率。
为了在AFE计算中达到收敛性和准确性,对所有相关构象进行采样非常重要。但是,由于不同构象之间的障碍很大,因此在典型AFE计算的时间尺度上,采样通常是不完整的。因此,结果通常取决于初始结构。已经开发了几种方法来增强采样。其中一种主要方法是将AFEM与副本交换方法(REM, replica exchange method )耦合。温度REM涉及并行运行具有不同温度的MD或MC模拟的副本,并在最近,人们开发了更有效的副本交换方法,并将其与FEP结合使用,即采用溶质回火(REST1和REST2,replica exchange with solute tempering)方法进行副本交换。对于标准FEP计算未捕获构象变化的系统,FEP / REST2已显示出可以解决采样问题并精确再现实验性ΔΔAs。另一种增强采样的方法是使用图形处理单元(GPU)。最近在GPU上实现了FEP,TI和MBAR,并且伴随着2个数量级的加速,可以实现更多采样。随着计算机和GPU的功能不断增强,可以对大型蛋白质配体数据集进行AFE计算快速且常规地执行
正在进行的第二项重大发展是在部队领域(force fields)。用于药物样有机分子的第一代力场是在1990年代末和2000年代初开发的,包括默克(Merck)大分子力场,OPLS,AMBER的GAFF和CHARMM的CGenFF。使用8个启用了GPU的FEP / REST2协议和OPLS2.1力场的FEP +程序(Schrödinger,Inc.)已针对8个蛋白质配体靶标进行了追溯验证。在八个系统上观察到的平均误差约为1 kcal / mol,并且据报道还为两个前瞻性药物设计项目可靠地预测了真正的阳性化合物。最近,GAFF v1.8力场与AMBER蛋白力场FF14SB结合在一起使用AMBER GPU TI实施针对同一数据集进行了验证。与实验相比,计算出的ΔΔ的RMSE(0.62-1.83 kcal / mol)与GPU FEP REST2用OPLS2.1力场进行计算,其中三个蛋白质配体系统除外,获得的RMSE(0.93-1.41 kcal / mol)相当。 对于GPU FEP / REST2计算,已经对扭转和共价参数进行了广泛的训练(针对10,000多种代表性有机化合物),因此部分假设AMBER GPU TI实施的性能较差是由于某些错误扭转参数。如前所述,关键扭转参数对FEP计算很重要。然而,在另一项报道的GAFF 1.8 / AMBERFF14SB力场组合的回顾性测试中,使用的计算协议与早期工作不同,最大RMSE从1.83降低至1.22 kcal / mol,尽管仅测试了八个蛋白质配体系统中的四个。在默克公司的最新报告中,他们认为,如果RMSE小于1.3 kcal / mol,并且离群值较大,则可以成功针对目标进行验证可以充分解释,并且所采用的力场/模拟设置适合将来针对同一目标进行前瞻性研究。根据这些现实标准,GAFF 1.8 / AMBERFF14SB组合和OPLS2.1力场均可用于针对所探究的八个目标中的大多数的前瞻性研究。但是,来自默克公司的同一项研究强调了确保AFEM能够常规获得高精度的许多其他代表性和技术问题。据报道,人们继续努力优化OPLS和AMBER / GAFF力场。通过重新拟合肽的二面体参数以及配体电荷模型的改进,Harder和合著者基于OPLS2.1力场开发了OPLS3力场。最近,对配体扭转类型的进一步广泛优化和配体部分电荷分配的优化导致了OPLS3e力场。在前八个蛋白质-配体数据集上使用GPU FEP计算对OPLS3和OPLS3e力场进行了测试,结果表明改进,RMSE分别为0.70-1.25 kcal / mol和0.63-1.17 kcal / mol。
GAFF2是基于GAFF开发的,范德华力,键,角度和二面体参数的重新参数化有望改善力场。最近,分别使用Flair中的MBAR和GROMACS,中具有pmx的非平衡TI,对具有AMBER蛋白力场的GAFF2(FF14SB和FF99SB-ILDN)针对大型数据集进行了测试。对于AMBER FF14SB / GAFF2和AMBER FF99SB-ILDN / GAFF2,先前提到的八个蛋白质-配体测试集的RMSE分别为0.29-1.68 kcal / mol和0.90-1.56 kcal / mol。还使用CGenFF v3.0.1和v4.1测试了非平衡TI方法,并且观察到了稍差的结果。提出了一种结合GAFF和CGenFF结果的共识方法,并发现获得与GAFF2类似的结果。结合其他一些数据集的结果,共识方法的总体MUE非常接近于使用FEP +的MUE,两者均约为0.87 kcal /mol。所有测试均表明,当前类药物分子力的性能领域与不同的AFEMs相结合对于回顾性研究是令人满意的;尽管对于某些蛋白质系统,与实验相比,RMSE需要降低,并且对于大多数测试,并未对整个数据集进行重复运行以获得严格的误差线。
现代力场使用原子类型描述不同化学环境中的不同原子,然后根据原子类型分配不同的非键合和键合参数。 CADD中AFEM的一个主要问题是,在药物开发项目中,类药物配体的化学空间很大,以至于现有的力场可能无法覆盖所有的配体原子类型。因此,需要进一步的原子类型和参数化工作来解决这一缺陷。最近,一种新的力场格式,即SMIRKS天然开放力场(SMIRNOFF)格式,被用于开发开放力场模型。 SMIRNOFF格式的力场参数是通过直接化学感知分配的,而不是基于原子类型的。新格式具有更大的灵活性和简便性,并且还可以避免基于原子类型的力场出现问题的情况。最近,将开放力场在再现QM分子结构和能量方面的性能与基于原子类型的力进行了比较。 另一个最近开发的力场,即量子机械定制(QUBE)力场也可以规避传统力场的参数可传递性问题。 QUBE力场从QM电子密度得出系统特定的非键合参数。它已被用于研究蛋白质-配体结合自由能以及蛋白质动力学,并显示出令人鼓舞的结果。
最后但并非最不重要的一点是,自动化是另一项重大发展。从历史上看,设置所有类型的自由能计算可能很棘手,耗时且容易出错,因此使这些方法更易于使用的关键步骤是简化设置,运行模拟然后分析结果。已经开发了许多工具,可以帮助自动执行AFE计算的不同阶段,从生成参数和拓扑的初始阶段到设置扰动路径。还有一些程序可以使整个AFE过程自动化。 使用这些工具和程序,可以以更快,更可靠的方式来进行大规模计算的设置和分析,这由于节省了时间,因此大大有利于在制药环境中部署AFEM。大型回顾性基准计算,例如使用FEP +,Flair,和pmx,进行的计算,也可以推动力场的提高。但是,尽管可以使用健全性检查来评估AFE计算的可靠性(例如,在Flair中),包括向前和向后扰动的滞后,周期闭合错误以及相邻的lambda窗口之间的重叠,但实际操作经验仍然发挥着重要作用。用户需要相关领域的专业知识和培训,例如,他/她对程序适用范围的理解,以确保正确使用部署了AFE技术的程序。此外,一切都自动化 ,用户将无法控制设置模拟详细信息,这可能会导致意外或意外的结果。
总体而言,在过去30多年的时间里,社区在部署AFEM方面的经验有起有落,但强烈感觉到,通过最终的推动,我们最终可能会拥有一种能够例行和可靠地加速药物开发的工具。最后的努力包括解决在工业环境中部署AFEM的一些关键的剩余挑战。在默克公司(Merck KGaA)关于AFEM实际使用的最新工作中,他们列出了在药物设计活动中使用AFEM时遇到的一系列问题。这些问题包括由于构象变化而导致的蛋白质结构的不确定性,配体结合模式的不确定性,X射线结构中缺失的成分,测定条件和模拟条件之间的差异,金属和辅因子以及两者的质子化状态的不确定性。蛋白质和配体。此外,对配体互变异构的正确处理仍然是一个挑战。此外,对于AFE计算,涉及电荷变化,支架变化,脂族链芳环等的扰动也带来了一系列挑战。
在CADD中使用AFEM的开发已经经历了30多年的激动人心的旅程,很有趣的是,随着计算能力的不断提高和新型分子表示形式的出现,未来几年AFE技术将如何发展。 一方面,在技术方面,未来看起来是光明的(例如更长的模拟,更好的力场等),但是处理系统设置的策略(例如质子化,原子丢失等)并不容易。 解决的问题,最终可能比纯粹的技术问题更具挑战性。
参考资料
- Evolution of Alchemical Free Energy Methods in Drug Discovery. Lin Frank Song and Kenneth M. Merz, Jr. https://dx.doi.org/10.1021/acs.jcim.0c00547
